
Optimizing FDA Submissions For Companion Diagnostics
By Janice Hogan, Partner and Co-Director of the FDA/Medical Device Practice, Hogan Lovells US LLP

By Janice Hogan and Shilpa Prem, Hogan Lovells US LLP
“Doctors have always recognized that every patient is unique, and doctors have always tried to tailor their treatments as best they can to individuals. You can match a blood transfusion to a blood type — that was an important discovery. What if matching a cancer cure to our genetic code was just as easy, just as standard? What if figuring out the right dose of medicine was as simple as taking our temperature?” — President Barack Obama, Jan. 30, 2015
In recent years, there has been significant progress in the development and understanding of the human genome, as well as the molecular diagnostics designed to facilitate “personalized medicine.” An increasing number of companion diagnostics are being developed to optimize matching between patients and specific therapies. With the proliferation of these new technologies, FDA has issued new guidance and continues to evolve its policies to support the advancement of these diagnostic tools.
In January 2015, President Obama launched the Precision Medicine Initiative (PMI) to promote the development of new, effective treatment and prevention strategies, targeted to patients based on their unique attributes. FDA has a significant role in this Initiative as the gatekeeper in making sure that such products are safe and effective.
In this article — part one of a two-part series — we will examine the history of companion diagnostic approvals by FDA. Through this process, trends can be identified to suggest better approaches to optimize review times.
Development, Review, And Approval Of Companion Diagnostics
Although FDA has been evaluating companion diagnostics and other personalized medicine technologies for over a decade, significant new guidance has been issued in the past two years. In August 2014, FDA issued Guidance for Industry: In Vitro Companion Diagnostic Devices – Final Guidance. This guidance replaced a 2005 concept paper and provides important insights regarding FDA’s recommended approach to developing companion diagnostics for both new and existing therapeutic products.
In the guidance, FDA explains that, ideally, a therapeutic product and a companion diagnostic would be developed contemporaneously. FDA recommends that sponsors developing companion diagnostics interact with FDA to obtain early feedback on plans for design and data development. While this is the ideal scenario in many respects — because it enables both simultaneous approval and a completely integrated development plan — the sequence of diagnostic versus therapeutic development may not fit this idealized scenario in many cases. Nonetheless, a number of companion diagnostics have been approved that were not contemporaneously developed.
To date, companion diagnostic devices and the corresponding therapeutic products have generally required separate FDA applications. A premarket approval (PMA) application, de novo application, or 510(k) notice may be required for the companion diagnostic device, depending on the level of risk, and a new drug application (NDA) or a biologic license application (BLA) will be required for the corresponding therapeutic product. Furthermore, each application may involve input from both FDA’s Center For Devices and Radiological Health (CDRH) and its Center For Drug Evaluation and Research (CDER), due to the collaborative nature of the review.
The regulatory pathway for clearance of a companion diagnostic will depend on the risk profile of the device. FDA stated in its Guidance that “experience indicates that most IVD companion diagnostic devices will be Class III devices, although there may be cases when a Class II classification with premarket notification (510(k)) is appropriate.” According to a list of all FDA-cleared and approved companion diagnostic devices, provided on the administration webpage, there were 27 PMA companion diagnostic approvals and one clearance via the de novo process between 2007 and 2015.
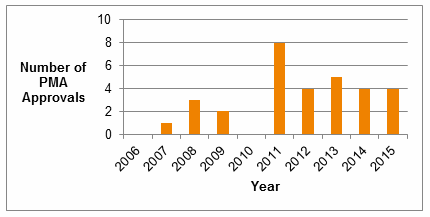
Fig. 1 – Number of PMA Approvals for Companion Diagnostics, 2006-2015
The review of companion diagnostics has in recent years been primarily assigned to the Division of Molecular Genetics and Pathology within CDRH. The creation of a specific staff, dedicated to personalized medicine products within CDRH, further illustrates FDA’s focus on this product category.
FDA also has focused on review times for companion diagnostics and, as noted by Dr. Jeffrey Shuren in his testimony before Congress, has met its review time performance goals for all recent companion diagnostics. Review times have steadily decreased over the past five years (Fig. 2), though submission volume for companion diagnostics in the past two years is relatively low.
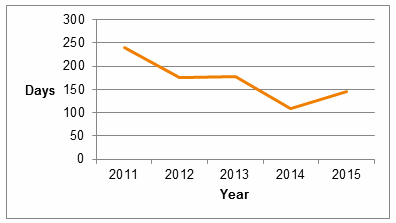
Fig. 2 – Average Review Time for Companion Diagnostic PMA Submissions
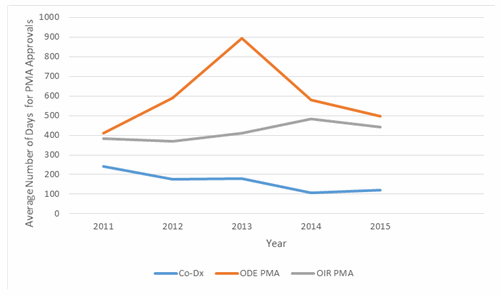
Considering only companion diagnostic products that required PMA approval, average review time was 146 days in 2015. This review time is well below the average for all PMA applications (Fig. 3). Notably, although PMA review times in the Office of In Vitro Diagnostics and Radiological Health (OIR) are typically shorter than the corresponding PMA review times for therapeutic products review in the Office of Device Evaluation (ODE), companion diagnostic products average significantly shorter review times than other products reviewed by OIR. These trends suggest a high degree of emphasis by FDA on prioritizing companion diagnostic review, and may also signify an effort to synchronize the timing of companion diagnostic approval with drug approval.
Data Requirements For Companion Diagnostic Approval
For companion diagnostics developed in parallel with a corresponding therapeutic agent, the amount of available data may be driven largely by the data needed to support drug approval. However, for companion diagnostics not developed in parallel with a drug, a variety of approaches have been taken to develop an adequate supporting data set.
As is the case with all diagnostic product development, FDA typically requires an initial testing phase for the companion diagnostic to finalize its operating characteristics, followed by a validation phase. For companion diagnostics, this can be done within the drug development process, for example, by developing the companion in phase II studies and validating it in phase III testing. Alternatively, if a diagnostic product is developed only after the therapeutic product, retrospective approaches may be taken to test validation.
The table included below this article provides a summary of the amount and type of data that has supported companion diagnostic approvals from 2013 to 2016 that are currently listed by FDA, the majority of which were primarily based on retrospective data. Approximately one-third of the companion diagnostic devices were approved simultaneously with either a drug or biologic.
In addition to its use of retrospective data, CDRH has established a strategic priority for 2016-2017 to increase the use of “real-world” clinical experience. By Dec. 31, 2017, the Center’s goal is to increase by 100 percent the number of premarket and postmarket regulatory decisions that leverage real-world evidence (compared to its FY2015 baseline). It is possible that this effort may offer another tool to evaluate companion diagnostic products, in either the premarket or postmarket settings.As indicated in the table, a number of the companion diagnostics were approved on the basis of bridging studies, which, for example, linked the results of a diagnostic test kit to earlier laboratory assessments or device versions used during the clinical study of the therapeutic. Recognizing that the timing for finalizing companion diagnostic tests may not always be “synchronized” with Phase III drug development, FDA has shown flexibility in allowing the use of bridging data to accommodate these types of development scenarios.
Conclusions
Since the first personalized medicine devices were developed, there has been significant progress in both the technology and the scientific and medical communities’ understanding of actionable genetic associations in various diseases. Correspondingly, complexity in the submission and review of companion diagnostics has increased.
FDA has been at the forefront of developing policies and guidance surrounding the development of personalized medicine products, and has made significant progress in decreasing companion diagnostic review times, with average PMA review times now consistently meeting or exceeding the Agency’s performance goals. The potential for single submissions of drugs developed simultaneously with companion diagnostics, as an alternative to the current dual submission process, may also help to simplify the regulatory process.
About The Authors
Janice Hogan is the co-director of Hogan Lovells' FDA/Medical Device practice. Janice focuses her practice primarily on the representation of medical device, pharmaceutical, and biological product manufacturers before the U.S. Food and Drug Administration (FDA).
A biomedical engineer, she held positions in marketing/marketing research for a major pharmaceutical manufacturer prior to becoming an attorney. She has authored articles regarding the medical device 510(k) review process, regulation of medical software, orphan drug regulation, use of foreign clinical data, the de novo review process, medical device products liability, and chapters of several textbooks related to medical device regulation.
Janice has served as an adjunct professor at the University of the Sciences in Philadelphia and as a guest lecturer at the Wharton School, Drexel University, and Stanford University on the development and regulation of medical products. She is also a frequent lecturer at FDA regulatory law symposia and conferences on topics related to premarket approval of medical products, combination products regulation, and product development. She has also served on the faculty of FDA's staff college training for new review staff.
Shilpa Prem is an associate at Hogan Lovells and represents medical device, pharmaceutical, and biological product manufacturers before the FDA. Shilpa has a biomedical engineering degree and has held various positions in large pharmaceutical and medical device companies.
Questions to the author may be directed to janice.hogan@hoganlovells.com.
|
Company/Device (Submission Number) |
Submission Type |
Summary of Clinical Studies |
Prospective vs. Retrospective Studies? |
Simultaneous Drug/Biologic Approval? |
|---|---|---|---|---|
|
ARUP Laboratories KIT D816V Mutation Detection by PCR for Gleevec Eligibility in Aggressive Systemic Mastocytosis (ASM) - H140006 |
HDE |
28 patients from phase II study and published reports |
Retrospective |
No |
|
ARUP Laboratories PDGFRB FISH for Gleevec Eligibility in Myelodysplastic Syndrome/Myeloproliferative Disease (MDS/MPD) - H140005 |
HDE |
31 patients from phase II study and published reports |
Retrospective |
No |
|
Resonance Health Analysis Systems Ferriscan R2-MRI Analysis System (DEN130012) |
De Novo |
Device used as part of the inclusion criteria and as the primary endpoint in the clinical studies (N=166, with long term follow-up in 133) for the use of deferasirox (Exjade) in the treatment of chronic iron overload with non-transfusion-dependent thalassemia syndromes (NTDT) |
Prospective |
|
|
Ventana Medical Systems, Inc., Ventana PD-L1 (SP142) Assay P160002 |
PMA |
Device was tested as part of a multicenter, open-label, two-cohort trial to evaluate atezolizumab. Patients had advanced or metastatic urothelial carcinoma. Tumor specimens from these patients were evaluated using device and results were used to define subgroups based on PD-L1 expression for pre-specified analysis (N=310). |
Prospective |
Yes |
|
Dako North America, Inc., PD-L1 IHC Nivolumab PharmDx P150027 |
PMA |
Device was tested as part of analysis of patient samples from Phase 3 study of nivolumab monotherapy or nivolumab in combination with ipilimumab versus ipilimumab monotherapy in subjects with previously untreated, unresectable or metastatic melanoma. (N=945). Patients were stratified in the drug study based on PD-L1 status express as assessed by the device. |
Retrospective |
No |
|
Dako North America, Inc., PD-LI IHC 28-8 pharmDX P150025 |
PMA |
Device was tested as part of analysis of patient samples from Phase 3 study of nivolumab vs docetaxel in adult (≥18 years) subjects with advanced or metastatic non-squamous cell NSCLC after failure of prior platinum doublet-based chemotherapy (N=455). Patients were stratified in the study based on PD-L1 result assessed by device. |
Retrospective |
No |
|
Abbott Molecular, Inc., VYSIS CLL FISH Probe Kit - P150041 |
PMA |
Device was tested as part of a Phase 2 study for the approval of venetoclax. Study required that a 17p deletion must have been documented using the device for enrollment in the study. The overall response rate data from this study was used to support approval of venetoclax. (N=261) |
Prospective |
Yes |
|
Roche Molecular Systems, Inc., cobas® EGFR Mutation Test – P120019 |
PMA |
Device was tested as part of the Phase 3 study via a bridging study to assess Tarceva (erlotinib). Device tested for the agreements between the device and the Clinical Trial Assay for detection of deletions of exon 19 and exon 21 mutations and clinical outcomes of patients enrolled in the overarching drug trial whose tumor specimens harbor exon 19 deletions or L858R mutations (N=134). |
Retrospective |
No |
|
Dako North America, Inc., PD-L1 IHC 22C3 Pharmdx – P150013 |
PMA |
Device was tested in analysis of samples from multicenter, open-label, randomized phase 1 clinical study conducted to assess the safety and efficacy of Keytruda (pembrolizumab) in patients with advanced non-small cell lung cancer (N=156). |
Retrospective |
No |
|
Qiagen Manchester LTD, Therascreen EGFR RGQ PCR Kit – P120022 |
PMA |
Device was tested as part of the Phase 3 study for afatinib and bridging study between the Clinical Trial Assay (CTA) and the device. To establish the clinical utility of device, clinical outcomes for all patients in drug trial were compared to the outcomes of patients whose specimens were mutation-positive upon retrospective testing of the device. (N=525) |
Retrospective |
Yes – PMA Supplement to extend label claim of the device to include an indication for Iressa (gefitinib) was approved at the same time as its associated NDA
|
|
Ventana Medical Systems, Inc., Ventana ALK (D5F3) CDX Assay – P140025 |
PMA |
Device was evaluated as part of a Bridging Study of a Phase 3 study of crizotinib vs. first-line chemotherapy in previously untreated patients with ALK positive locally advanced or metastatic nonsquamous NSCLC. Objective was to determine concordance between the device and Vysis ALK Break Apart FISH Probe Kit assay. (N=1099) |
Retrospective |
No |
|
Roche Molecular Systems, Inc., cobas KRAS Mutation Test – P140023 |
PMA |
Study compared the device with Sanger sequencing and the FDA approved QIAGEN therascreen KRAS RGQ PCR Kit for detection of mutations in codons 12 and 13 of the KRAS gene in CRC tumor specimens using a subset of subjects enrolled in the phase 3 trial comparing capecitabine plus oxaliplatin with bolus fluorouracil/leucovorin as adjuvant therapy for Stage III colon cancer. (N=461) |
Retrospective |
No |
|
Myriad Genetic Laboratories, BRACAnalysis CDx – P140020 |
PMA |
The device was tested as part of a Bridging study of a clinical study that assessed olaparib treatment in patients with ovarian cancer who have a deleterious or suspected deleterious germline BRCA mutation and who have been previously treated with at least three lines of prior chemotherapy. Bridging study assessed agreement between the device and the local test results for gBRCAm detection. |
Retrospective |
Yes
|
|
Qiagen Manchester Ltd., therascreen KRAS RGQ PCR Kit – P110027 |
PMA |
Device was tested as part of Phase 3 clinical study. Banked tumor samples from patients in Phase 3 study were tested with the device to identify 1) KRAS mutation-positive (mutant) and no mutation detected. (N=1100) |
Retrospective |
No |
|
BioMerieux, Inc., BioMerieux THxID BRAF Assay Kit – P120014 |
PMA |
Device was tested as part of Phase 3 clinical study. Bridging study was done to evaluate 1) the agreement between the THxID-BRAF Kit and the CTA for detection of BRAF V600E mutations and 2) to assess the clinical outcome of the patients enrolled in the overarching clinical trial whose tumor specimens were V600E positive as detected by the device. (N=584) |
Retrospective |
Yes |