
World Health Organization (WHO) Unveils Global Regulatory Framework For Medical Devices

By Jof Enriquez,
Follow me on Twitter @jofenriq
The World Health Organization (WHO) has released a draft proposal for its global regulatory framework for medical devices and in vitro diagnostics (IVDs).
A WHO working group started to develop its model for regulating medical devices in 2015, based on an agency resolution that called for the establishment and strengthening of “regional and subregional areas of regulation of health products that are the least developed, such as regulation of medical devices including diagnostics.”
According to RAPS, most countries have a national medicines regulatory authority and regulatory standards for pharmaceuticals but absent or limited regulatory systems for medical devices and IVDs. Because of a dearth of financial or technical resources, it is challenging for many countries to transition successfully from an unregulated market to a comprehensive medical devices law in a single program. Instead, each country will have to take a different approach to building its own regulatory systems for devices. WHO offers their framework under development as a model for WHO member states as they take steps to ensure the quality and safety of medical devices available in their jurisdictions.
The WHO Global Model Regulatory Framework for Medical Devices Including IVDs is based on standards developed by the Global Harmonization Task Force (GHTF) and its successor, the International Medical Device Regulators Forum (IMDRF).
The framework provides guidance for a staged development from developing law with basic regulatory requirements, to registration of establishments that put medical devices on the market, to listing of medical devices and post-market controls.
In the document, WHO recommends that medical devices and IVDs may be grouped into risk classes according to their potential for harm to the patient or user, using a widely accepted general classification and associated documents made by the GHTF.
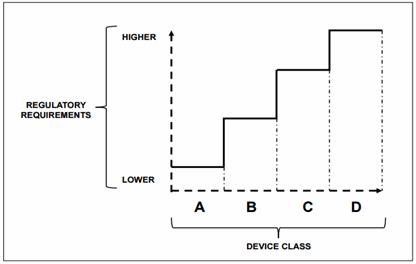
Instead of mentioning specifics, WHO lists “essential principles” that device manufacturers must demonstrate to a country’s regulator that a medical device has been designed and manufactured to be safe and perform as intended during its lifetime. For regulators, WHO offers “principles of good regulatory practice” and “essential tools for regulation” but does not provide detailed, country-specific guidance. It does, however, provide references to relevant documents on the topic of regulation.
WHO asserts that “medical device regulation must have a sound basis in law” and recommends countries to enact legislation that “delineate the responsibilities of the regulatory authority and establish its enforcement powers to include removing products from the market as well as consequences for bad practices,” and to “establish the responsibilities of manufacturers, importers, distributors and authorized representatives.” It recommends authorities conduct a gap analysis of existing controls to assess the degree to which national regulations are aligned with international guidance and best practices, and to build regulatory capacity and resources required. Sample questions of the gap analysis could include:
- Are medical devices regulated at all?
- Are they currently regulated as medicines or some other product category?
- Is there a specific and sound legal foundation for regulation of medical devices?
- What is the public health risk in the country associated with medical devices?
- Is there a national regulatory authority with clear powers and responsibilities for medical devices?
- What proportion of medical devices are imported and from where?
- Are there local manufacturers of medical devices? If so, are their activities regulated and how?
The WHO framework recommends a “progressive” or “stepwise” approach to regulating the quality, safety, and performance of medical devices from basic controls to expanded controls. It also describes circumstances in which a regulatory authority may rely upon or recognize the work products from trusted regulatory systems (scientific assessments, audit, and inspection reports) or WHO prequalification.
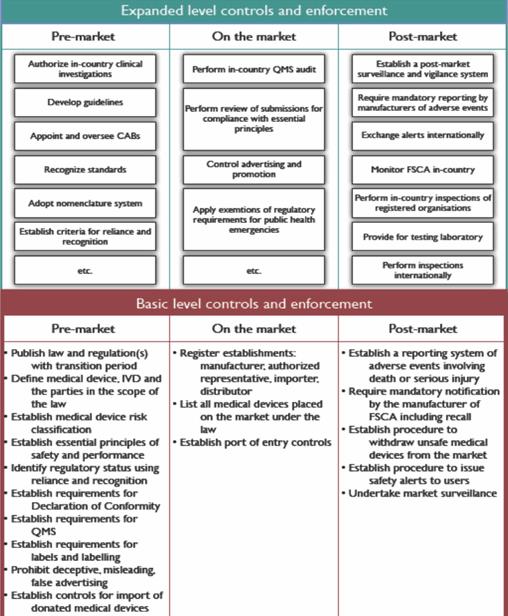
WHO clarifies that their framework provides a general approach rather than specific recommendations for a country’s national authority tasked to construct or run an effective system regulating medical devices and IVDs. It does not detail responsibilities of other stakeholders such as manufacturers, distributors, and health-care professionals, all of whom have roles in assuring the quality, safety, and performance of medical devices.
Public comments on the draft proposal will be accepted until June 20, 2016.