
Implementing UDI Direct Marking

Direct marking is one part of FDA’s Unique Device Identification (UDI) rule, and it has a set of compliance dates that may differ from the labeling and Global UDI Database (GUDID) submission dates. The recently published draft guidance document Unique Device Identification: Direct Marking of Devices restates some requirements for direct marking and clarifies some issues. The real advantage is that it brings all of this information together in one place.
The UDI system has three major elements: adding a UDI to the product , loading a large dataset to FDA’s public database (the GUDID), and updating the quality system procedures.
Direct marking primarily falls into the product element, which requires a UDI on the device label and on the label of every standard packaging configuration. In some cases, the product itself must have the UDI, (i.e., the device is directly marked). GUDID has data elements related to direct marking.
Direct Marking Requirement
Direct marking means permanently adding the UDI to the device. The idea is that a device will be in use for a long time, become separated from its label and, as a result, the UDI would be lost. The solution is to put the UDI on the device itself.
The UDI rule defines when a device must have direct marking. The basic requirement involves three factors: the UDI is required on the device label; the device is intended to be used more than once; and the device is reprocessed before each use.
Direct marking generally applies to all device classes, but there is one exception. A Class I device that has a UPC on the label and device packages is exempt from the direct marking requirement [see 21 CFR §820.40(d)].
Direct Marking Dates
The UDI rule is phased in over time, so there is a schedule for direct marking. The UDI system utilizes compliance dates for the various elements. The table below shows the direct marking compliance dates. An applicable, finished device manufactured and labeled on or after the listed compliance date must have direct marking.

For a finished device manufactured and labeled before the compliance date, there is a grace period of three years from the compliance date. For example, consider a life-sustaining or life-supporting device manufactured and labeled on June 1, 2015. The direct marking compliance date is Sept. 24, 2015, so this device would not require direct marking until Sept. 24, 2018. However, a life-sustaining or life-supporting device manufactured and labeled on Oct. 1, 2015 would require direct marking, and the three-year grace period would not apply.
Direct Marking Format
The UDI rule does not require a specific format for direct marking, and it provides device makers some options. One of the factors is that the device must have a UDI on its label. The full UDI on the label is the device identifier (DI) and any optional production identifiers (PI). The requirement is that the direct marked UDI convey the same information. For example, if the label UDI includes a serial number, then the direct mark UDI also must include the serial number.
The first option is the device identifier (DI). A device manufacturer could distinguish between a packaged device and an unpackaged device by using a different DI. For example, the manufacturer may want to distinguish between complaint records for the packaged device and for the unpackaged device. Since complaint investigation records must contain the UDI, per §820.198(e)(3), a different DI could help.
The second option is the presentation of the information. The label requires the UDI to be easily readable in both plain-text and in automatic identification and data capture (AIDC). However, the direct marking could be one or the other or both. The manufacturer decides.
For easily readable plain-text, the UDI rule prescribes the format, while the issuing agency’s standards apply to AIDC.
Understanding Reprocessing
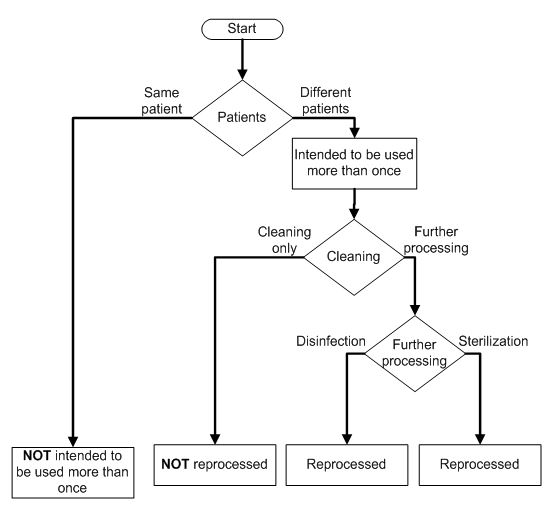
A device that is intended to be used more than once and reprocessed between uses requires direct marking. The UDI rule doesn’t define these terms, but the draft guidance provides working definitions that clarify the meaning: A device “intended to be used more than once” means the same device is used again and again on different patients.
This raises the issues of carryover and contamination, which reprocessing addresses. Reprocessing is the mechanism by which a previously used or contaminated device is made suitable for the next use. Reprocessing is intended to remove blood, tissue, biological debris, etc., and to inactivate infectious microbes, thereby making the device safe for the next patient.
In clarifying reprocessing, the direct marking guidance refers to another guidance document, Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling. The flowchart below helps determine whether direct marking is required. In this case, reprocessing means:
- Clean and disinfect before the next use on a different patient; or
- Clean and sterilize before the next use on a different patient.
If the device only is cleaned between different users, it does not require direct marking UDI. Similarly, if the device is intended to be used more than once for the same patient, it does not require direct marking UDI.
How to determine if a device is reprocessed:
Direct Marking Exceptions
There are some cases where the manufacturer can take an exception to direct marking. Evaluate the device and its intended use to determine if any of these exceptions apply:
- Any type of direct marking would interfere with the safety and effectiveness of the device,
- It is not technologically feasible to directly mark the device,
- The device is cleared/approved as a single-use device, or
- The device already has a permanent UDI directly marked on the device.
If the manufacturer wishes to take an exception, it must document the reason in the device design history file, §820.30(j). The manufacturer does not request the exemption from FDA. In loading GUDID, check the box “Device Subject to Direct Marking (DM), but Exempt.”
Safety And Effectiveness
Adding direct marking to a device is a design change that must implement §820.30(i). One major question is the effect of direct marking on the safety and effectiveness of the device. The design change analysis should identify any areas of concern. If any type of direct marking would interfere with the device’s safety and effectiveness, the manufacturer can document an exception and provide the decision to GUDID.
There could be cases when the manufacturer decides to mark the device anyway. For example, a major hospital chain might require direct marking on certain types of devices. Where there is an impact on safety and effectiveness, FDA requires a regulatory submission. For a 510(k) or de novo device, this means a new 510(k) submission. For a PMA or BLA device, this means a supplement.
In both cases, 510(k) and supplement, the applicable user fees apply. There are no user fee waivers for submissions associated with direct marking.
Moving Forward
With clarifications to the draft guidance document, a manufacturer should move forward on the direct marking portion of UDI. The first compliance date is Sept. 24, 2015!
Start by understanding how your device fits into the UDI system. First, determine if FDA-CDRH considers the device as life-sustaining or life-support. Enter the product code into the Classification Database. The result will tell you if the device is life-sustaining/life-supporting and it will tell you the device class.
Determine if the three attributes requiring direct marking apply to your device:
- The device label must have a UDI.
- The device is intended for more than use.
- The device is reprocessed between uses.
To help answer these questions, follow the draft guidance and the flowchart above. You probably will need to review the instructions for use, any marketing information, and the clearance or approval letter.
Using the decision on direct marking, the life-sustaining/life supporting information, and the device class, determine the compliance date from the table provided above.
Initiate the design-change process required by §820.30(i). Part of the process will determine if an exception is warranted or not. Hold a design review before the compliance date and, in the minutes, include the agenda item and document the decision.
If the decision is for direct marking, be sure to validate the permanence for the anticipated number of uses and subsequent reprocessing.