
The What, Why, When, And How Of Risk Management For Medical Device Manufacturers
By Robert Di Tullio, Senior VP, Global Regulatory Services, Beaufort
Over the years, the discipline of quality in the medical device industry has developed from a reactive practice to one of ensuring a total quality approach throughout a product’s lifecycle.
My first introduction to a proactive quality, rather than reactive “quality control” program, approach was in the 1970s, not in the medical device field but rather the automotive industry. At that time, the United States was under siege by imports, especially those from Japanese carmakers. The public began to turn to these imports more and more, due to improved quality rather than U.S. “planned obsolescence.”
In response, the quality field began to reinvent itself using continuous quality improvement approaches such as Six Sigma, Kaizen, and others — which crossed all company departmental boundaries, rather than focusing only on the quality organization. At the heart of effective proactive quality practices is root cause analysis and associated methods that form the basis for risk management. These practices have evolved into polished disciplines during the ensuing decades.
This article will explain the value of a robust risk management program for medical device manufacturers by briefly describing the what, why, when, and how of risk management.
What Is Risk Management?
Risk management is a systematic application of management policies, procedures, and practices to the tasks of analyzing, evaluating, controlling, and monitoring risk. The International Standards Organization (ISO) Technical Committee 210 (ISO/TC 210), Quality management and corresponding general aspects for medical devices, was formed in 1994, and the group published the first edition of the ISO 14971 risk management standard for medical devices in 1998. A second edition of this standard was published in October 2007 as ISO 14971:2007, Medical devices - Application of risk management to medical devices. The standard describes the requirements for risk management to determine the safety of a medical device by the manufacturer during the product life cycle.
Why Should You Care About Risk Management?
A risk management program is required by higher-level regulation and other quality standards. ISO 13485, the standard that represents the requirements for a comprehensive quality management system for the design and manufacture of medical devices, states under Product Realization: “the organization shall establish requirements for risk management throughout product realization.” Risk management is also a requirement of the FDA’s Quality System Regulation (QSR), especially under 21 CFR 820.30 Design controls (g) Design validation: “Design validation shall include software validation and risk analysis, where appropriate.” This standard, adopted by European Committee for Standardization (CEN) as EN ISO 14971:2012, is also harmonized with respect to the current European Medical Devices Directive 93/42/EEC, and is required by ANSI/AAMI/IEC/EN 60601-1-2:2014 Medical electrical equipment – Part 1-2: General requirements for basic safety and essential performance, subclause 4.2. In addition, it is recommended by the International Medical Device Regulators Forum (IMDRF), which supplanted the Global Harmonization Task Force (GHTF/SG3/N15R8) in 2011.
Although there are myriad regulatory reasons to perform risk management, the most important reason is plain and simple: It is integral to designing quality and safety into a product.
When Should You Perform Risk Management?
I am often asked by clients, “When is the right time to perform risk management?” My answer is to start at the beginning and never cease — that is, you should perform risk management continuously throughout the product lifecycle. For example, risk management is critical:
- During the initial design of the product, to assure that potential hazards and defects are recognized and designed out,
- With each design modification, to revisit original assumptions and to assure that any changes “do no harm” to that original product,
- With each production process change, to ensure that improvements intended by lean manufacturing or other concepts to reduce scrap and/or cost, etc. are completely thought through prior to initiating them, and
- After each product recall. Yes problems occur, even with the most robustly designed products, leading to recalls to remove violative or unsafe products from the hands of users. When these unfortunate events occur, a smart company will revisit the risk analysis file to critique whether such hazards may have been foreseen with a rigorous and objective risk management effort.
Again, risk management starts at the beginning and must never cease.
How Do You Perform Risk Management?
In my estimation, the how is both the most difficult and most enjoyable part of risk management. ISO 14971 allows for many options and approaches to performing risk management, and each organization should choose that one that best fits its culture. Some examples include:
- Fault tree analysis (FTA), a top-down approach
- Failure modes and effects analysis (FMEA) and failure modes, effects, and criticality analysis (FMECA), both bottom-up approaches
Modified versions of these methods are acceptable according to the standard and are actually my favorites. A modified FMECA that employs a three-tiered quantitative scoring system for estimation of hazards and risks for each hazard works best, in my experience. But again, I encourage you to find the optimal approach that best fits your organization.
Most importantly, the risk management approach you choose must be taken very seriously and objectively, or it will produce nothing but useless paper in a technical file. A robust program begins with highly qualified personnel on the risk management team who develop a risk management plan and file. The actual risk analysis process must take into account the intended use and characteristics related to the safety of the device, and only a well-qualified and objective team can focus within this context.
The team should begin with a thorough identification of all potential and/or known hazards and develop an estimation of the risk(s) for each hazard. In my three-tiered version, the severity, probability of occurrence, and detectability of each hazard are evaluated, scored, and placed in a hierarchy of “danger of risk” using a cutoff value to identify and assign risk mitigation candidates. One can even choose to employ a qualitative version of this, depicted in Figure 1 below.
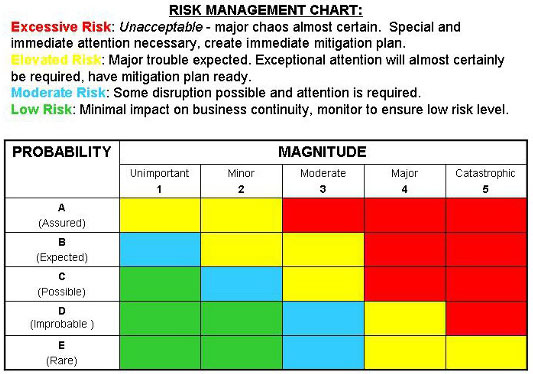
Figure 1
Once this evaluation is complete, the team should then focus on risk control and risk reduction and implementation of risk control measure(s), sometimes taking into account risk/benefit analysis and risks arising from risk control measures. The team completes its effort by publishing a risk management report to be used as production and post-production information throughout the product lifecycle. This goes hand-in-hand with the widely accepted PDCA (plan-do-check-act) practices shown in Figure 2 below.
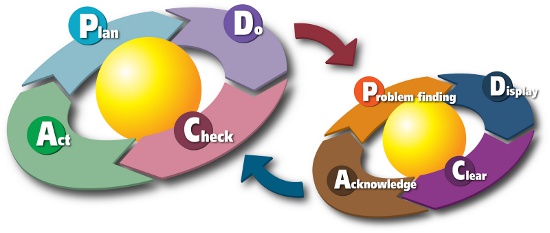
Figure 2
In closing, my advice is to take your risk management process very seriously and perform it thoroughly — it can and will prevent failure, problems, and compliance nightmares from occurring.
 About The Author
About The Author
Robert Di Tullio is an independent consultant in medical device regulatory, quality systems, clinical affairs, and healthcare public policy, specializing in diagnostics. For the past 40 years, he has held various positions with multiple organizations, with a particular emphasis on quality and regulatory management. Visit www.ditullioconsulting.com to learn more.