
A Brief Guide To ISO 13485's Design Controls In Medical Device Development
By Thom Wyatt, Principal Engineer, Springboard

The development of medical devices requires meticulous attention to detail and a focus on ensuring patient safety. Design controls play a crucial role in the development process by providing a systematic approach to manage risks, design requirements, and verification and validation activities, and ensuring that devices are designed, developed, and manufactured in a consistent and controlled manner. By implementing robust design controls, developers can:
- Enhance patient safety: Design controls enable you to identify and mitigate risks associated with the use of medical devices, reducing the potential for harm to patients and users.
- Improve product quality: Effective design controls help to establish and maintain quality throughout the development process, resulting in reliable and consistent devices that meet user needs and regulatory requirements.
- Facilitate regulatory compliance: Compliance with design control requirements is a critical aspect of obtaining and maintaining regulatory approvals and certifications, such as the CE mark or FDA clearance.
The internationally recognized standard that outlines the requirements for design controls in the medical device industry is ISO 13485, which in this aspect is in alignment with the U.S. rules for design controls in FDA 21 CFR Part 820.30. Let’s explore the key elements of ISO 13485 related to medical device development.
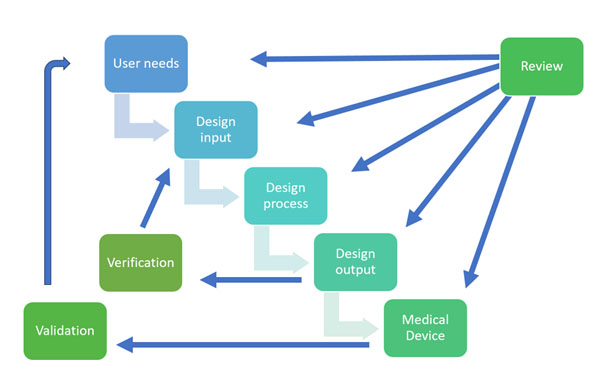
Design And Development Planning: ISO 13485 emphasizes the importance of planning the design and development process. It helps to create a comprehensive design and development plan that outlines the activities, tasks, and responsibilities involved in developing the medical device. This plan should address design inputs, outputs, verification, validation, and the overall development process, which includes development stages and determining the necessary resources and responsibilities.
Design Inputs: Design inputs specify the requirements that a medical device must meet to address the needs of users, patients, and other stakeholders. These inputs include performance requirements, safety and regulatory requirements, and user needs. This includes features, performance characteristics, materials, and any other applicable specifications.
Design Outputs: Design outputs are the tangible results of the design process, such as drawings, specifications, and manufacturing instructions. ISO 13485 requires that these outputs be documented and reviewed to ensure they meet the design inputs and are suitable for manufacturing, testing, and quality control processes.
Design Reviews: Regular design reviews are conducted throughout the development process to assess the progress, identify issues, and verify that the design outputs meet the design inputs. These reviews involve cross-functional teams and provide opportunities for feedback and improvement on the designs and devices.
Design Verification: Perform testing and analysis to ensure that the design outputs meet the design input requirements. This includes activities like inspections, laboratory testing, and performance evaluations. It’s important to also clearly document the results of these verification activities to show due diligence and proof of process.
Design Validation: Validate the medical device design to ensure it meets the intended use and user needs. This may involve clinical evaluations, usability studies, and other relevant assessments. Once again, documenting the validation activities and results is an important part of the process.
Design Transfer To Manufacture: Design transfer involves the transfer of the design to manufacturing, ensuring that all necessary information and documentation are provided to facilitate production. ISO 13485 emphasizes the importance of effective communication and collaboration between design and manufacturing teams during this phase. Without this, there could be delays and higher costs due to lack of the correct and detailed information transfer.
Production And Quality Control: Establish manufacturing processes, quality control procedures, and inspection methods to ensure consistent production of the medical device in accordance with the design specifications. This ensures consistency for the production and easy methods to check quality control once manufacturing has begun.
Design Changes: The standard requires a systematic approach to managing design changes, including documenting, reviewing, and approving any modifications made to the device design. All change reviews must evaluate the effect on existing products, risk management, and product realization activities. This is where, if the process has been followed, time and costs for design changes can be minimal. If the processes have been implemented correctly, then design iterations can be concluded faster, allowing for shorter delays in the long run.
Design And Development File: The conformity to these design and development requirements needs to be recorded for each medical device. This can be in a dedicated file or as references to existing records. In the U.S., these files are known as Design History Files. For regulatory compliance, prepare and compile the required technical documentation, including a risk management file, design history file, and instructions for use. Other applicable regulations may need to be considered if selling into different target markets around the world.
Design controls are an integral part of medical device development, contributing to the safety, quality, and regulatory compliance of devices. ISO 13485 guidelines for design controls provide a comprehensive framework to manage the design and development process effectively. By following these controls, we can mitigate risks, improve product quality, and ultimately deliver safe and effective medical devices to improve patient care.
 About The Author:
About The Author:
Thom Wyatt is a principal engineer at Springboard. He is experienced in the design and development of high-risk medical devices.