
A Guide To Device, Label, And Package Requirements Of The UDI Rule
By Dan O'Leary, President, Ombu Enterprises

On the surface, unique device identification (UDI) is a simple concept. Put the device identifier (DI) on the device label in machine‑readable form. Include the production identifiers (PI) that you have selected, and make sure they are both machine‑readable and human‑readable. Ensure the format for any human‑readable dates are in the required format. Load the data into the FDA-CDRH Global Unique Device Identification Database (GUDID), and update the quality systems procedures. However, many complexities add complications.
Accredited Agency
The device manufacturer does not assign the device identifier (DI). Instead, it comes from an organization accredited by FDA. These organizations are called accredited agencies or, because they issue the DI, issuing agencies. To date, FDA has accredited three agencies: GS1, Health Industry Business Communications Council (HIBCC), and ICCBBA.
Note that the accredited agency you select is a supplier that provides a service. Be sure you have implemented the requirement, evaluation, and selection process in 21 CFR 820.50.
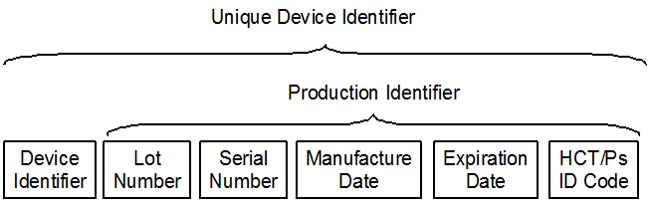
Figure 1: The components of a UDI
Each of these organizations has a required format for the UDI. You can view the details in the document UDI formats by FDA-Accredited Issuing Agency [Word doc]. For the DI the data has the following format. There are also similar format requirements for the PIs.
GS1 14 numeric characters
HIBCC 6 to 23 alphanumeric characters
ICCBBA 18 alphanumeric characters
In addition to the data structure, the accredited agencies may allow (or restrict) the data carrier. Generally, the data carrier implements automatic identification and data capture (AIDC). The data carrier is the method used to present the machine‑readable information. Allowed data carriers include linear bar codes, 2D bar codes, QR codes, RFID chips, etc. FDA does not specify the data carrier; the device manufacturer decides based on what customers can read and what the accredited agency allows. For example, a manufacturer could use an RFID chip. However, if the customer cannot read it, there is little value in this approach. It appears that the vast majority of manufacturers will implement linear bar codes.
The Date Format
The rule requires that human‑readable dates conform to a standard format to avoid confusion and misleading users. The standard format, in most cases, uses year-month-day (e.g., 2013-09-30). The exception is the date of manufacture for electronic products that FDA regulates. In 21 CFR 1010.3(a)(2)(ii) the required date format is the month without abbreviation and the year as a four-digit number (e.g., Manufactured: September 2013).
One important point is that the AIDC may include dates using characters. For example, there are often printed characters under a linear bar code. These dates are part of the AIDC as specified by the issuing agency. FDA does not regulate how the AIDC communicates data; these dates do not have to be in the human‑readable date format.
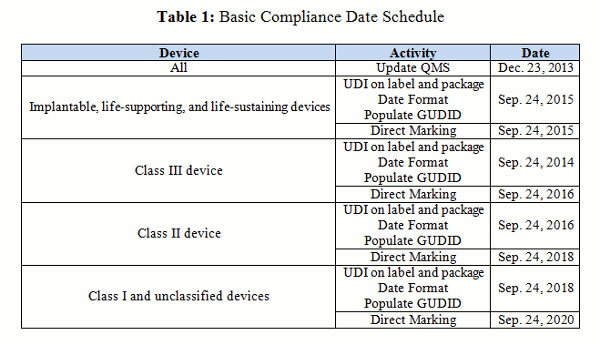
The date format goes into effect on the compliance date (see Table 1).
The Base Package
In the simplest case, there is one device in a package with a label. This is the base package. The label has the UDI (DI + PI), correct date formats, etc.
The DI for the base package is the key to the GUDID record, which contains the identification information for that device.
There are a variety of DIs in the system as shown in Figure 2. Each one has a particular application.
In some cases, a device manufacturer may have more than one accredited agency for a device, meaning that the device will have more than one DI. In these instances, the manufacturer designates one as the primary DI and the other as the secondary DI. The distinction is included in GUDID.
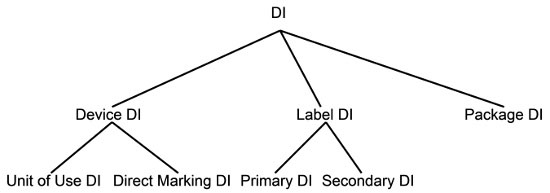
Figure 2: A taxonomy of device identifiers (DIs)
Packaging Configurations
The easiest way to understand packaging configurations is to think about inventory management at the customer’s warehouse. With UDI in place, the customer would like to scan the label on a box on the shelf and determine the contents. For example, does the box have only one device or five devices inside? A new DI for the box solves the problem.
The device has a UDI = DI + PI, on the label of the base package. If the device is available in a box of, say, five, then the box label has a DI linked to the device label DI. The manufacturer populates the GUDID with the package DI and the quantity per package.
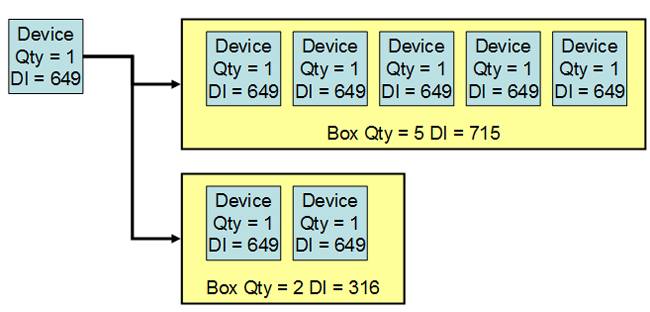
Figure 3: Each packaging configuration has a different device identifier (DI)
One decision for the manufacturer is the PIs to use for a packaging configuration. Because the PIs are optional, the device label and packaging configuration label could have different PIs. For example, the package may always contain devices from the same lot, so the lot number at the packaging configuration would be appropriate. If the packaging configuration has different serial numbers, then the serial number would not be in the package UDI, but would be in the device label UDI.
Direct Marking
In some instances, the UDI is on the device itself, which is called direct marking. Some devices will be in use long after they come out of the package. Without the package, the device will not identify itself. The solution is to mark the device. The marking should be able to survive normal use, cleaning, and processing. The direct marking compliance date is two years after the base package compliance date.
A device requires direct marking when it meets three conditions:
- The device is intended to be used more than once.
- The device is intended to be processed before each use.
- The device must have a UDI on its label.
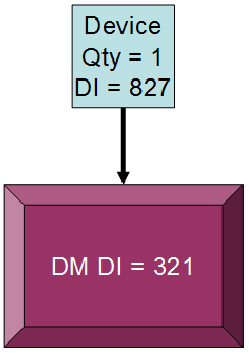
Figure 4: Using a direct marking device identifier (DI)
In some cases, direct marking may present a technical problem. The manufacturer can take an exception when direct marking meets any one of the following conditions:
- Direct marking would interfere with the safety or effectiveness of the device.
- It is not technologically feasible.
- The device is a single-use device subject to additional processing for an additional single use.
- The device has been previously marked.
The manufacturer must document the exception in the design history file [21 CFR 820.30(j)].
The direct marking DI could be the same as on the base package or different to distinguish it from the packaged device. The manufacturer decides, and includes the decision in GUDID.
Special Cases
The regulations provide for some special cases briefly described below.
Stand‑Alone Software
Stand‑alone software is itself a device, in contrast with software built into a device. There are two basic methods to distribute stand‑alone software: packaged form or downloaded.
When distributed in packaged form, the label and device package must also include a UDI in both plain text and AIDC.
When not distributed in packaged form, the software must display a plain text statement of the UDI whenever the software is started, or a plain text statement displayed through a menu command (e.g., an "About . . ." command).
The compliance date for stand-alone software matches the device class or the life-supporting or life-sustaining date (see Table 1).
Unit Of Use
A base package usually contains only one device. However, in some instances there may be more than one. For example, a box may contain 100 examination gloves or 25 adhesive bandages. The individual glove or bandage will not have a UDI, but there will be a UDI on the box. A hospital, for example, needs to distinguish between using one glove on a patient and using a box of 100. To solve this problem, the manufacturer assigns a “unit of use” DI and loads it into GUDID so that it links to the base package DI.
Kits
A convenience kit is multiple different medical devices packaged together for the convenience of the user. This could include procedure trays, IVD kits, etc.
The convenience kit requires a UDI on the kit’s label. However, the individual devices inside the kit do not need a UDI. The UDI on the kit label is sufficient. For example, a surgical procedure tray would require sterilization, but a UDI on the items in the tray could interfere with sterilization of the devices in the kit.
If the device in the kit were sold separately (perhaps as a replacement), then it would require a UDI on the base package label.
Combination Products
A combination product, in this case, is a device with a pharmaceutical or biologic component.
- For a single entity combination product, they are physically, chemically, or otherwise combined or mixed and produced as a single entity.
- For a co-packaged combination product, they are packaged together in a single package or as a unit.
A single entity combination product with a National Drug Code (NDC) number on its label does not require a UDI.
The device constituent of a co-packaged combination product must have a UDI on its label. There is an exception for a UDI on the constituent device, if there is a UDI on the combination product. This means the combination product would have both an NDC number and a UDI.
Class I Devices
Class I devices are the lowest risk class in the FDA hierarchy. Consequently, there are some special conditions for UDI.
The UDI of a Class I device is not required to include PIs.
A Class I device with a universal product code (UPC) on the label and packages meets the UDI requirements. This means the UPC is the DI. The manufacturer must provide all the GUDID information.
Some Class I devices are of such low risk that FDA exempts them from the 21 CFR 820, except for some record keeping requirements. These devices do not require a UDI on the label or package. They are also exempt from providing data to GUDID.