
A Regulatory Perspective: FDA Final Guidance For Design Changes Requiring New 510(k) Submissions
By Jeff Ryberg, Regulatory Affairs Professionals Society (RAPS)
 Submissions")
In response to technological advancements, broadening medical knowledge, and clinical use, medical device designs frequently evolve and come in many forms. Designs often change in response to industry pressure for increased customer satisfaction and device safety, maximized performance, maximized manufacturability, and cost efficiencies.
Regulatory professionals within the medical device industry have many roles — one of which is the impact assessment of proposed design and labeling changes on devices that previously received marketing clearance from the FDA or a pre-amendment to a device that is subject to 510(k) requirements. A new premarket notification (510(k) filing) on a medical device is required for "a significantly changed or modified design, components, method of manufacture or intended use" per U.S. Federal Regulation 21 CFR 807.81 (a)(3).1
Industry Outlook
Global medical device sales are estimated to increase by 6.4 percent annually from 2016 to 2020, reaching nearly $440 billion.2 While the Americas are projected to remain the world’s largest medical device market, the Asia-Pacific and Western European markets are expected to expand at a quicker pace over the next several years.3
In 2011, the medical device market in the EU accounted for one-third of the global market, with around $122.5 billion in yearly revenue.4 The U.S. is expected to continue to play a leading role in medical device R&D. After declining in 2009, R&D spending rebounded to $2.9 billion in 2010 and $7.3 billion in 2011. From 2013 to 2020, larger medical device companies are expected to increase their R&D budgets by approximately three percent, while the rest of the industry is expected to increase spending for this element by more than five percent.5
In the U.S., for a manufacturer to register and list a device as part of 21CFR Part 807 requires the submission and clearance of a premarket notification 510(k), or the device cannot be commercially distributed legally. In an industry experiencing such growth, it is important for manufacturers to understand these 510(k) regulatory changes and implement revised SOPs to avoid misalignment with the FDA further down the line as the device design evolves.
Design Changes And 510(k) Filing
In August 2016, the FDA posted a revision of the guidance Deciding When to Submit a 510(k) for a Change to an Existing Device, which superseded the version that was introduced in 1997.6 In the new draft guidance, the FDA sought to provide clear definitions of key terms in highly subjective criteria that have given rise to differences in interpretation and application of the guidance, such as "could significantly affect the safety or effectiveness" and "major change or modification."
Medical device and healthcare organizations are at significant risk of repercussions if the FDA and industry reach a conflicting 510(k) filing decision. When the industry determines a new 510(k) filing is not necessary for a proposed design or labelling change, it documents the non-filling decision, but it is still subject to review and scrutiny by the FDA at future site inspections. An FDA investigator might disagree with a company's non-filing decision, which could result in the investigator citing the firm for failing to meet 21 CFR 807.81 and issuing an inspectional observation (Form 483). The organization can then be issued a warning letter or product recall if they fail to adequately address the noted deficiency. Other areas of misalignment between the FDA and industry regarding the 1997 guidance include:
- lack of current, pertinent examples7
- disconnect between the flowcharts and supporting text8
- lack of correlation to risk analysis
- absence of clear documentation expectations.
With the issuance of final guidance documents in October 2017, the FDA realized its commitments to Congress by "preserving the basic format and content of the original, 1997" guidance and remained consistent with the 2016 draft guidance. In the final guidance, the FDA expanded on the guiding principles and labeling requirements and added more clarity and examples. The 10 new "guiding principles" have a significant impact on the process, decisions, and documentation required. They are summarized below in Table 1.

Device Modification Guidance: Key Changes
Section A: Labeling Changes — and the associated flowchart — represents the most significant change between the final issue and 2016 draft guidance and increase the scope. The first question in A1 changed from a “substantive change to indications for use” to “a change to indications for use.” The FDA expanded and clarified that all indications for use changes are in the scope, not just those deemed by the manufacturer as substantive.
The other significant change and increased scope is labeling section A4: “does the change affect the instructions for use” to “could the change affect instructions for use.” The FDA again opens the scope to include any possible effect of the change, not just those known to affect change.
Usability and over-the-counter clarification questions were also added. The FDA increased the number of labeling examples in Appendix A from eight to 11, addressing industry comments and requests.
Sections B, C, D, and E remain primarily unchanged from the draft.
Final Software Guidance
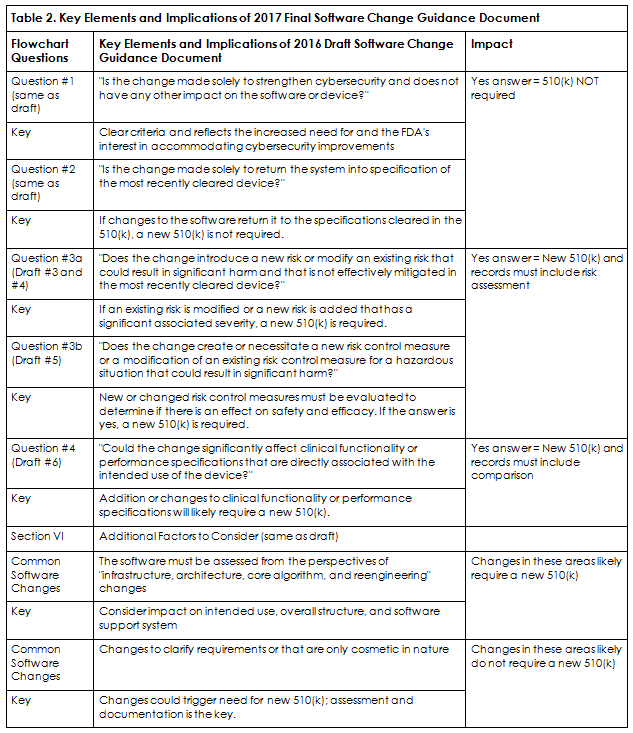
The final software guidance primarily follows the 2016 draft, with additional clarification and a streamlined flowchart. The FDA considers all software changes to be design changes by definition and added additional emphasis in the final guidance regarding what could significantly affect safety and effectiveness. Table 2 is an example flowchart for organizations to follow and specifies when a software change requires a new 510(k) submission. The following questions lead the assessor to a defensible software change 510(k) filing decision. Note: question 3a and 3b both need to be answered.
Recommendations
The FDA has clarified the significant role of risk management in the regulatory filing decision process. The burden is on the industry to adopt and implement an effective risk-based change assessment process for its filing decisions. Several recommendations for updating existing regulatory filing decisions and processes related to design and intended use changes are provided below.
First, include an application/documentation of a risk-based assessment linked to your risk management process to substantiate your decisions.
It also is recommended that a comparison of the proposed changes against the cleared device be available and to include current and aggregate changes since device clearance. Furthermore, software change assessment documentation must address the questions in Table 2.
It is recommended you look at your label review processes and the resultant documentation requirements and ensure they are robust and cover the full “new” scope of the guidance.
Key functional stakeholders should be able to provide expert input to address clinical, design, and risk management considerations to ensure a fully comprehensive and error-proof decision process. Trial runs before implementing any changes are advisable. This is particularly important for smaller organizations and those with infrequent design changes.
Further considerations include the addition of a functional "swim lane" into the decision-process flowchart so there is full clarity on role responsibility and deliverables, and flexible forms/templates (similar to the FDA's Appendix B) to ensure full process scalability.
Final Thoughts
To comply with the final guidance, revised procedures, associated documentation, and implementation into the quality management system will require cross-functional planning and execution as well as an increase in the time and resources required. However, the FDA anticipates that because the new guidance documents only add clarification, the impact of these new procedures on organizations will be negligible. Whether this is true and if there is an increase in 510(k) submissions as a result of the change remains to be seen.
References
- Title 21: Food and Drugs. Part 807—Establishment Registration and Device Listing for Manufacturers and Initial Importers of Devices. Subpart E-Premarket Notification Procedures. GPO website. http://www.ecfr.gov/cgi-bin/text-idx?SID=f35b20cdc0edc588fb4fb00139c86b60&mc=true&node=se21.8.807_181&rgn=div8. Accessed March 14, 2017.
- https://www.trade.gov/topmarkets/pdf/Medical_Devices_Top_Markets_Report.pdf
- http://mercercapital.com/article/5-trends-to-watch-in-the-medical-device-industry-in-2016/
- https://www.emergogroup.com/resources/market-europe
- The State of the U.S. Medtech Industry, January 2015, Anchin, Block & Anchin
- Deciding When to Submit a 510(k) for a Change to an Existing Device K97-1 Memorandum and Guidance Issued January 10, 1997.
- FDA's CDRH Webinar entitled: Deciding When to Submit a 510(k) for a Change to an Existing Device Draft Guidance for Industry and FDA Staff and Deciding When to Submit a 510(k) for a Software Change to an Existing Device Draft Guidance for Industry and FDA Staff. August 2016.
- Inspection Observations. FDA website. https://www.fda.gov/ICECI/Inspections/ucm250720.htm. Accessed March 14, 2017.
About The Author
Jeff Ryberg is an active enterprise member of the Regulatory Affairs Professionals Society (RAPS) and holds the position of vice president, regulatory affairs and quality assurance, Danaher Business System Integration and Special Projects for Danaher Corporation. Ryberg has 30 years of experience in medical device quality and regulatory affairs and supports Danaher's diagnostics, life sciences, dental, and water technologies businesses. He has an M.S. in regulatory science from Johns Hopkins.
Join RAPS in Dublin April 24 to 25, 2018, for an exclusive two-day workshop: The Basics of 510(k) and Working With FDA.