
An Introduction To International Medical Device Standards
By Dan O'Leary, President, Ombu Enterprises

One common source of misunderstanding in the medical device industry is the method the various national regulatory systems use to identify standards. This article explains the method, starting with standards from the International Organization for Standardization (ISO) adopted and recognized in various regulatory systems. The article uses ISO 13485:2003 and ISO 14971:2007 as illustrations.
International Standards
There are two organizations that typically issue international standards: the International Organization for Standardization (ISO) and the International Electrotechnical Commission (IEC).
These standards are international standards, meaning they apply to the world. Consequently, any given region or country could adopt them, perhaps with modifications or limitations.
International standards are denoted, typically, with three parts. First is the issuing organization, second is a number, and third is the year of issue. For example, ISO 14971:2007 is an international standard that ISO issued in 2007. The title is Medical devices — Application of risk management to medical devices.
Other examples are:
- ISO 13485:2003, Medical devices — Quality management systems — Requirements for regulatory purposes
- ISO 10993-1:2009, Biological evaluation of medical devices — Part 1: Evaluation and testing within a risk management process
- IEC 62366-1:2015, Medical devices — Part 1: Application of usability engineering to medical devices
On occasion, ISO will issue technical reports related to specific standards. These are often considered guidance documents that help the reader implement the standard. For our primary examples, ISO has issued ISO/TR 14969:2004, Medical devices — Quality management systems — Guidance on the application of ISO 13485:2003, and ISO/TR 24971:2013, Medical devices — Guidance on the application of ISO 14971.
On occasion, an ISO standard needs a correction, but the correction isn’t significant enough to warrant creation of a new version of the standard. One example is International Standard ISO 13485:2003 Technical Corrigendum 1, published in 2009 to correct some typographical errors.
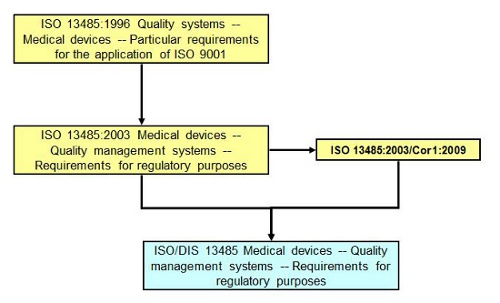
History of ISO 13485:2003
An international standard doesn’t “know” where it operates. For example, ISO 13485:2003 has many notes relating to “national or regional regulations.” These are instances where a country (such as Canada) or a region (such as the European Union) may adopt regulations that differ from or clarify specific clauses of ISO 13485:2003.
In citing an international standard, it is best to include the three elements (organization, number, and year) to avoid ambiguity.
Adoption
A region or a country has a standards organization that may adopt the international standard, and in some cases they modify it or place limitations on it. In addition, the medical device authorities in the region or country may recognize the standard, but there is no obligation to do so. We will use the U.S., EU, and Canada to illustrate these points.
United States
In the U.S., the standards organization is the American National Standards Institute (ANSI). It is the U.S. representative to ISO. ANSI is composed of other U.S. organizations that may become involved in adopting American national standards. Two important organizations in this area are the Association for the Advancement of Medical Instrumentation (AAMI) and the American Society for Quality (ASQ).
When ANSI adopts a standard, the organizations involved show up as a prefix. In the U.S. our example standards are:
- ANSI/AAMI/ISO 13485:2003 (R2009), Medical devices — Quality management systems - Requirements for regulatory purposes
- ANSI/AAMI/ISO 14971:2007 (R2010), Medical devices — Application of risk management to medical devices
ANSI has a policy to review standards on a regular basis. If the standard is reaffirmed, that date, with an “R”, follows the initial date.
Another well-known standard is ANSI/ISO/ASQ Q9001-2008: Quality management systems — Requirements, which is the U.S. version of ISO 9001:2008.
The U.S. standards organization may adopt a standard, but the U.S. medical device regulatory authority, the Food and Drug Administration (FDA), doesn’t have to recognize it. The FDA maintains a list of recognized consensus standards for medical devices. That list includes:
- ISO 14971:2007, Medical devices — Application of risk management to medical devices
- ANSI/AAMI/ISO 14971:2007 (R2010), Medical devices - Application of risk management to medical devices
Significantly, it does not include either the international or U.S. versions of 13485, because FDA requires a different quality management system for medical devices marketed in the U.S.
European Union
The EU has two standards organization that are relevant in this case: the European Committee for Standardization (CEN) and the European Committee for Electrotechnical Standardization (CENELEC). Think of CEN as the EU organization that corresponds with ISO, and CENELEC as corresponding with IEC.
When CEN issues a standard based on an ISO standard, it adds the prefix “EN” and adjusts the year accordingly. In the EU our example standards are:
- EN ISO 13485:2012, Medical devices — Quality management systems — Requirements for regulatory purposes
- EN ISO 14971:2012, Medical devices — Application of risk management to medical devices
CEN issues standards but does not sell them. Copies come through designated sales points, which are the national standards organizations in the EU member states. Each of these standards organizations adds its own prefix. For our examples, the United Kingdom versions are available from BSI (British Standards Institution) as British standards and become:
- BS EN ISO 13485:2012
- BS EN ISO 14971:2012
Similarly, for Ireland NSAI (National Standards Authority of Ireland) offers:
- I.S. EN ISO 13485:2012
- I.S. EN ISO 14971:2012
The EU has a system of product directives that define the essential requirements for the particular types of products. In this case, the applicable directives are the Medical Device Directive (MDD), Active Implantable Medical Device Directive (AIMDD), and the In Vitro Diagnostic Directive (IVDD). Some standards have special status relative to the directives — they are harmonized. Harmonization offers a legal “presumption of conformity,” meaning that implementation of the harmonized standard shows the device conforms to that part of the Essential Requirements in the directive.
A standard becomes harmonized to a directive after publication in the Official Journal of the European Union. In addition, the EU maintains an informative website for each product directive listing the harmonized standards.
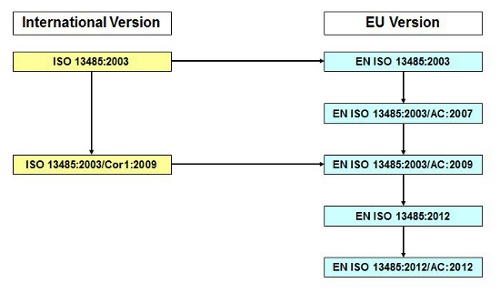
History of EN ISO 13485:2012
The EU states that the product directives for medical devices are more restrictive than the international standards. Consequently, the EU versions explain why following the international standards will not satisfy the product directives.
Canada
In Canada, the standards organization is the Canadian Standards Authority (CSA). For our example standards, the Canadian versions are:
- CAN/CSA-ISO 13485-03 (R2013), Medical Devices — Quality Management Systems — Requirements for Regulatory Purposes
- CAN/CSA-ISO 14971-07 (R2012), Medical Devices — Application of Risk Management to Medical Devices
The regulatory authority for medical devices is Health Canada, which publishes a list of recognized standards for medical devices.
The recognized standards, based on our examples, are:
- CSA-ISO 14971-07, Medical devices — Application of risk management to medical devices
- ISO 14971:2007, Medical devices — Application of risk management to medical devices
For most medical devices sold in Canada, the manufacturer must have a certificate stating that the quality management system under which the device is manufactured satisfies National Standard of Canada CAN/CSA-ISO 13485:03, Medical devices — Quality management systems — Requirements for regulatory purposes.
Summary
Standards are easy to understand if you keep a few simple concepts in mind.\
First, remember the system of prefixes. If you mention a standard, be sure to include the correct prefixes. They change depending on the regional or national application.
Second, remember to include the year, since it is a significant portion of the designation.
Third, standards may change based on the national or regional regulations. For example, in the EU we know that ISO 14971:2007 does not satisfy the product directives; device manufacturers implement EN ISO 14971:2012.
Fourth, countries or regions don’t adopt standards from another country or region. For example, the U.S. will not adopt EN ISO 14971:2012. One reason is that it includes specific requirements from the EU product directives that don’t apply to devices marketed in the U.S.
Image credit: Image credit: "Flags in front of UN Headquarters" by USAID. Licensed under CC BY 2.0 via Flickr.