
Are The Special And Abbreviated 510(k) Clearance Mechanisms Still Viable?
By Janice Hogan, Partner and Co-Director of the FDA/Medical Device Practice, Hogan Lovells US LLP

In March of 1998, FDA issued The New 510(k) Paradigm - Alternate Approaches to Demonstrating Substantial Equivalence in Premarket Notifications - Final Guidance.1 The guidance offered two alternative mechanisms to a traditional 510(k) notice, each designed to reduce regulatory burden and, consequently, review time. The first, the special 510(k) notice, was designed to allow a 30-day review, rather than the standard 90-day review, for changes to a device that did not alter the indications for use or fundamental scientific technology, compared to the manufacturer’s own previously cleared device. The second, the abbreviated 510(k) notice, was designed to allow reliance on guidance documents, special controls, or consensus standards to streamline the 510(k) submission and allow for more expeditious review.
In the years since these alternative 510(k) mechanisms were created, the extent of their use has changed, as has FDA’s approach to reviewing these types of submissions. Particularly with the introduction of FDA’s Refuse to Accept (RTA) 510(k) checklists in December 2012 — updated in August 2015 — the scope of submissions eligible for these alternative mechanisms has been clarified and potentially modified.2
For example, the special 510(k) RTA checklist stipulates that a submission can only be accepted as a special 510(k) notice if no performance test reports are provided.3 The RTA checklist states that “if performance data are provided and are conducted under design validation (21 CFR 820.30(g)), for example, to demonstrate continued conformance with a special control or recognized standard, then a Special 510(k) may be appropriate.”4 However, FDA’s application of this requirement since implementation of the new checklist indicates that the inclusion of performance test reports, with limited exceptions (e.g., software validation reports), will typically result in rejection of a special 510(k) during the RTA review, thus requiring submission of a new, traditional 510(k).
To assess whether FDA’s interpretation of the special and abbreviated 510(k) mechanisms has changed since these procedures were initially introduced, the 510(k) clearances in each category have been reviewed over time and by review division. Average review time for each submission type has also been evaluated to assess whether these mechanisms remain viable and beneficial.
The Use Of Both Special And Abbreviated 510(k)s Has Decreased Substantially
After the special 510(k) and abbreviated 510(k) were introduced in 1998, their use gradually increased as device manufacturers became more familiar with these mechanisms. Each type of 510(k) submission’s annual number of clearances is illustrated in Figure 1.
Figure 1 — Number of Traditional, Special, And Abbreviated 510(k) Notices Cleared Annually, 1998-20145
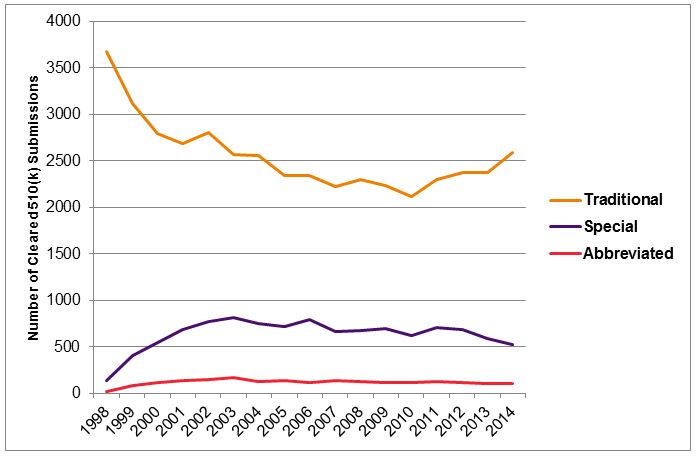
Use of the special 510(k) process peaked in 2003, when 819 special 510(k)s were cleared. But, just 520 special 510(k) notices were cleared in 2014, a decrease of over 35 percent. The number of abbreviated 510(k) notices cleared also peaked in 2003, at 173 submissions, compared to 106 in 2014, nearly a 40 percent decrease. Over the same period, the number of traditional 510(k)s cleared annually has remained relatively constant at approximately 2,500. Thus, there is a clear trend of decreased use of these alternative mechanisms over time. The implementation of updated RTA checklists in 2013 appears to have further reduced use of the special 510(k), with a decrease of approximately 25 percent between the number of special 510(k)s cleared in 2011-2012 and those cleared in 2013-2014, as shown in Figure 2.
Figure 2 — Number Of Special 510(k) Notices Cleared, Pre- Vs. Post-RTA Checklist6
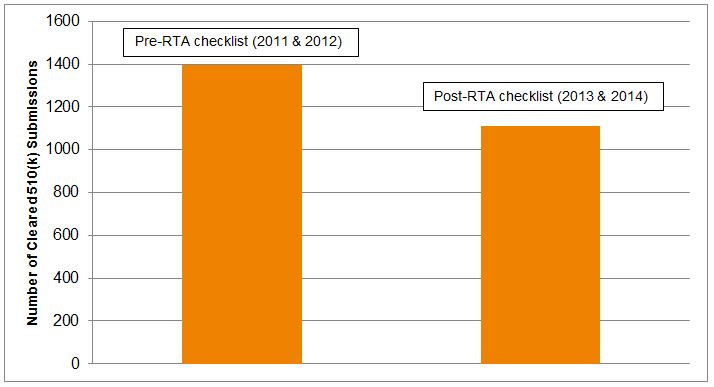
Data suggests that these trends are continuing, with fewer than 450 special 510(k) notices and only 52 abbreviated 510(k) notices cleared through mid-November, 2015.
The Effectiveness Of Alternative 510(k) Mechanisms In Reducing Review Times Is Decreasing
As noted above, the purpose of The New 510(k) Paradigm guidance and the development of the special and abbreviated 510(k) mechanisms was to reduce review times and regulatory burden on manufacturers. However, in recent years, the proportion of alternative 510(k) submissions with meaningful reduction in review times was limited. Of the special 510(k) submissions that were cleared in 2014, excluding any submissions rejected during RTA review, only approximately 36 percent were cleared in 30 days or less.
Thus, even though the number of special 510(k) notices has decreased significantly, and FDA appears to be screening special 510(k) notices more critically to ensure that they qualify for this mechanism, there has not been a corresponding improvement in review time for special 510(k)s. Similarly, for abbreviated 510(k) notices, only 18 percent were cleared in 60 days or less in 2014.
Average review time for special 510(k) notices in 2014 was 69 days. Over time, the review period has steadily increased for all types of 510(k)s, as shown Figure 3.
Figure 3 — Average Review Time For Alternative 510(k) Submissions Versus Traditional 510(k) Notices, 1998-20147
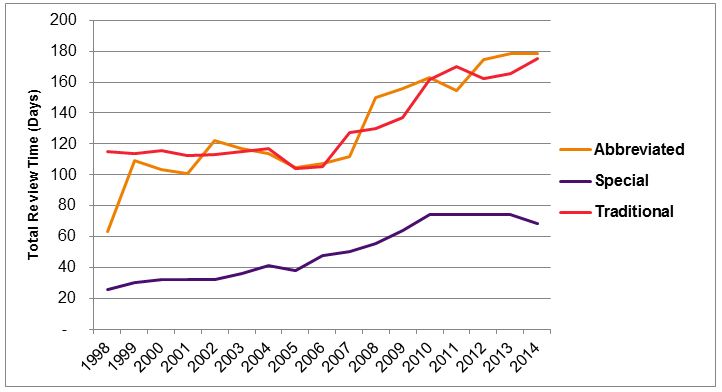
Average review time for abbreviated 510(k) notices in 2014 was 178 days, compared to 175 days for traditional 510(k) notices. Thus, there is no evidence of a review time advantage for abbreviated 510(k) notices. Review times for 2015 (to date) are similar, with an average review time of 161 days for abbreviated 510(k) notices and 165 days for traditional 510(k) notices.
Use And Effectiveness Of Alternative 510(k) Mechanisms Vary Significantly Across Therapeutic Categories
Use of special and abbreviated 510(k)s appears to vary by review branch, with greater use of these mechanisms in certain therapeutic areas, as shown in Figure 4.
Figure 4 — Proportion Of Special 510(k)s By Reviewing Branch, 20148
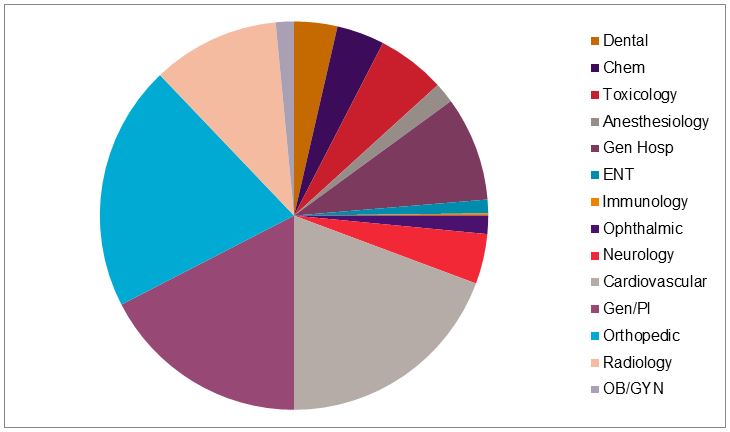
Among those branches that cleared more than 50 special 510(k) notices in 2014, use of the special versus traditional 510(k) mechanisms varied significantly. Approximately 25 percent of the cardiovascular 510(k) notices were special 510(k)s, compared to only 10 percent of all obstetrics and gynecology 510(k)s.
Including the use of both special and abbreviated 510(k)s, the trends were similar, with the greatest use of these mechanisms — as a percent of all 510(k)s reviewed — represented by cardiovascular products, followed by general and plastic surgery products, and then orthopedic products. No branch used the abbreviated 510(k) mechanism to a great extent, but dental and radiology products comprised the largest number of abbreviated 510(k)s reviewed.
Figure 5 — Percent Of 510(k) Submissions Reviewed In 2014 As Special Or Abbreviated, By Therapeutic Category9
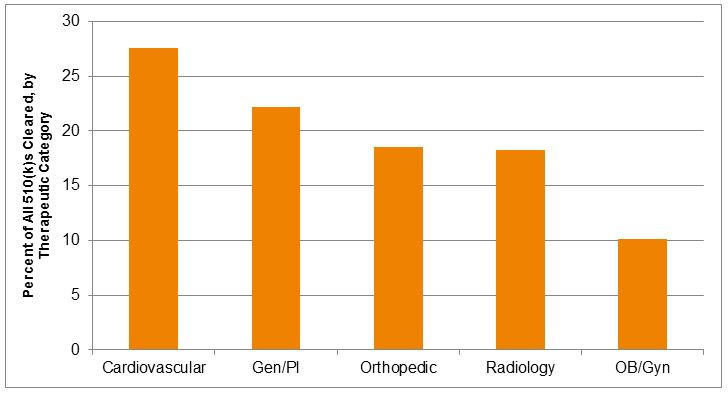
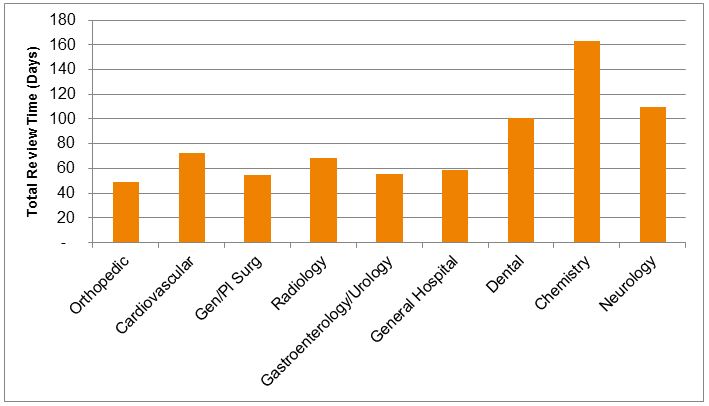
Review time performance also varies significantly by therapeutic category. While generally, those groups that review the largest number of special 510(k)s show shorter review times, there is still substantial variability. Orthopedic special 510(k)s had the shortest mean review times in 2014 (49 days), compared to longer review times in cardiovascular (73 days) and radiology (69 days). Overall, the review times for special 510(k)s for in vitro diagnostic devices were generally longer than for therapeutic devices. Although the number of special 510(k)s for diagnostics was relatively low, most categories of diagnostic products showed mean special 510(k) review times in excess of 100 days, suggesting relatively little advantage over typical review times for traditional diagnostic product 510(k) notices.
Figure 6 — Mean Review Time For Special 510(k) Notices in 2014, By Therapeutic Category (Days From Submission To Clearance)10
Conclusions
Since the special and abbreviated 510(k) mechanisms were created in 1998, the use of these alternatives to traditional 510(k) submissions has evolved. Although no clear policy change has been articulated by FDA that would account for these trends, it appears that these changes may reflect less willingness on the part of the FDA to clear 510(k) submissions without review of the underlying performance test data. Permissible use of both special and abbreviated 510(k)s has been narrowed by FDA, particularly under the RTA procedures instituted in 2013.
While special 510(k) notices may offer the potential for reduced review time in therapeutic areas where this type of 510(k) submission is common (e.g., orthopedics), the use of special 510(k)s is relatively infrequent and less advantageous in other therapeutic areas. In certain categories, such as most in vitro diagnostics, the average review time for special 510(k) notices is significantly longer than the intended 30 days, often longer than 100 days, and therefore offers less potential compared to traditional 510(k) notices.
The abbreviated 510(k) mechanism is used relatively rarely, representing a very small fraction of all 510(k)s submitted annually. Most therapeutic areas review less than 10 abbreviated 510(k) notices per year, with some exceptions (e.g., dental devices).
Special 510(k)s continue to offer shorter mean review times compared to traditional 510(k)s, but the reported statistics vary considerably by therapeutic area and do not include the effect of special 510(k) rejection, requiring conversion to a traditional 510(k). Given the risk of rejection during a special 510(k) notice’s RTA review period, the benefits of pursuing a special 510(k) are diminishing over time in most therapeutic categories. In addition, the advantage of pursuing an abbreviated 510(k) notice appears to be negligible in reducing review times, compared to traditional 510(k) submissions. Sponsors should critically evaluate these mechanisms and consider limiting their use to therapeutic areas where there is evidence of improved review times.
About The Author
Janice Hogan is the managing partner of Hogan Lovells' Philadelphia office and is co-director of its FDA/Medical Device practice. Janice focuses her practice primarily on the representation of medical device, pharmaceutical, and biological product manufacturers before the U.S. Food and Drug Administration (FDA).
A biomedical engineer, she held positions in marketing/marketing research for a major pharmaceutical manufacturer prior to becoming an attorney. She has authored articles regarding the medical device 510(k) review process, regulation of medical software, orphan drug regulation, use of foreign clinical data, the de novo review process, medical device products liability, and chapters of several textbooks related to medical device regulation.
Janice has served as an adjunct professor at the University of the Sciences in Philadelphia and as a guest lecturer at the Wharton School, Drexel University, and Stanford University on the development and regulation of medical products. She is also a frequent lecturer at FDA regulatory law symposia and conferences on topics related to premarket approval of medical products, combination products regulation, and product development. She has also served on the faculty of FDA's staff college training for new review staff.
Questions to the author may be directed to janice.hogan@hoganlovells.com.
Resources
- Accessed at http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm080187.htm.
- Accessed at http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm315014.pdf.
- Id.
- Id. at Appendix C, page 67 of 76.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm. Therapeutic categories were assigned based on review panel.
- Data accessed at http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.