
Are You Really Ready For FDA's Upcoming UDI Deadlines? Practical Tips For Compliance Success
By Tom Beatty, Sr., Principal, UDI Operations, QDevice (A Division of QPharma, Inc.)
Several months have passed since the release of the UDI final rule, and the clock is now ticking. Milestone dates are starting to loom large for device makers. The dust has settled a bit, and companies are (or should be) applying resources and building plans to reach compliance. The purpose of this article is to bring you up to speed on the latest UDI developments, and to point out some things you may want to think about as you build your UDI compliance programs.
Timelines Are Now Firm — No Longer Conjecture
While UDI compliance timelines were sketched out in the draft rule, the final rule locked the schedule down. It also offered an option for Class III device makers to formally request an extension of the first compliance date. At the UDI Conference in Baltimore, when this extension possibility was first revealed, there was an audible sigh of relief from the audience.
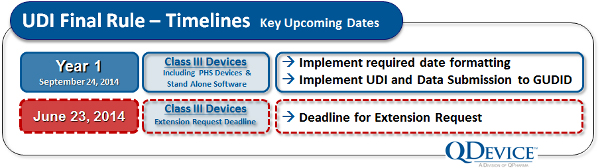
The FDA was very quick to explain, however, that extensions would only be granted in cases that clearly were in the best interest of the public health, and that extension requests would be scrutinized carefully. More or less, they were saying that they were willing to consider extension requests for specific devices, but that an extension is by no means automatic — there needs to be a clear and compelling reason for each extension given.
Our advice: If you think you will have trouble meeting the compliance date for Class III devices, begin assembling your extension request immediately, and start collecting data and other evidence to build a public-health-oriented rationale for extension.
For Class III devices (where an extension request has not been made), there are two key activities that must be completed by September 24, 2014. First, the UDI labeling must appear on any device sold. Second, the Global UDI Database (GUDID) submission must be complete. The GUDID (pronounced “good ID”) is FDA’s master database that will serve as the publicly accessible nexus of information related to approved medical devices.
To ensure that all goes smoothly, it is imperative to have GUDID data for your Class III device accurately assembled and submitted well in advance of the September 24 deadline. Submitting your device data earlier than the required compliance date will leave time to address any errors and avoid any potential volume bottlenecks in the submission process as the compliance deadline nears.
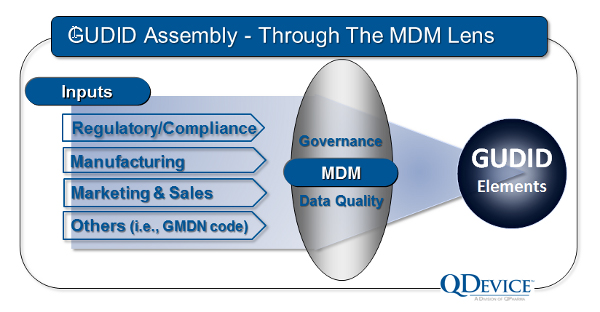
GUDID Required Elements
If you are a Class III device maker, chances are that you are well on your way to assembling the required data elements for your device’s GUDID submission. If not, you must begin immediately, as assembly of these fields can be a complex exercise.
It’s not too early for Class II and Class I device manufacturers to begin GUDID assembly, as well — and to pay attention to any lessons learned about the submission process following the compliance deadline for Class III devices.
So what are the key data elements? As per Appendix B of the GUDID Draft Guidance for Industry, there are 62 elements in 6 categories:

Clearly, this is an extensive list. And just to clarify a point, the production information (PI) fields — in the dashed boxes in the above illustration — will only be submitted as “flags” to show that the UDI will carry that information. It is not necessary to submit PI data to the GUDID database after each production run.
Assembling this data and its configuration for fluent submission to the GUDID will not be an easy or simple task. A device’s single GUDID submission may require the pulling of data from a variety of sources and functions in your enterprise. And remember that, after approval, the above data fields will be fully readable and searchable by healthcare providers and the general public, so accuracy is both required and vital.
GUDID Submission — Getting The Flow Right
The GUDID is planned to function for devices in the same manner as the National Drug Code (NDC) Directory does for pharmaceutical products. Using lessons learned from the NDC Database, FDA has built in some flexibility for GUDID submission. While accurate and successful GUDID submission is the goal, tuning the flow of many sources of data can really help the staging and assembly of the device’s UDI required data elements. Here are some tips to help ensure a smooth process:
Look at your big picture
In our experience gained from helping companies get ready for UDI compliance, it helps to take a big-picture view of how UDI-related data elements flow through your organization. In the figure below, we show an example schematic of the flow of information related to a device’s GUDID record.
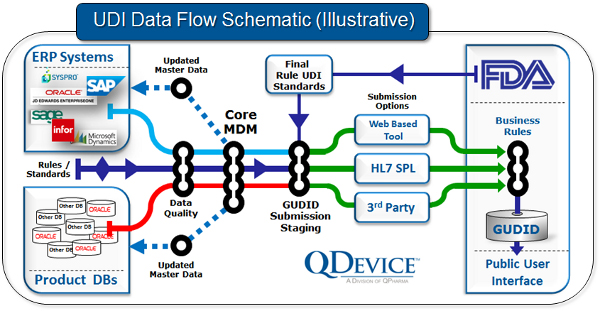
Use your MDM investment
If your organization has an existing or nascent master data management (MDM) program, it can be a powerful tool to speed GUDID data collection. The required element data fields require accurate and consistent data assembly from start of manufacture to use by the patient. GUDID submission requirements can be a powerful lens to focus and align resources to improve your MDM program and bring benefits across your organization.
Consider external resources
Rather than devote full-time resources to the assembly of records for GUDID submission, many companies are using external resources that have experience in pulling these fields together. Chances are, the external experts have already worked through the challenges of pulling GUDID fields from multiple systems and functions, and can accelerate the process cost-effectively.
Think about how you plan to submit
To drill down into our flow diagram, there are basically three options for uploading your device’s record into the GUDID:
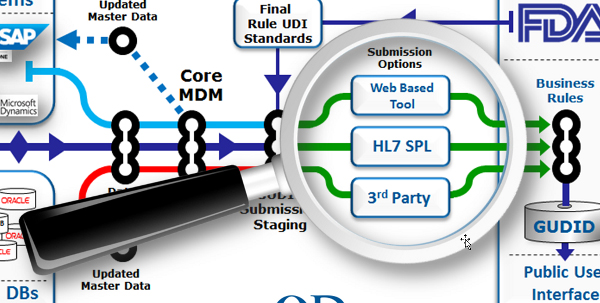
- Web-based submission: This option is primarily aimed at smaller manufacturers with a smaller number of SKUs for which they need to submit GUDID records. The key positive attribute here is a browser-based solution that can be accessed easily with a minimum of special training. The drawback is that with web-based submissions there is always potential for latencies and interruptions in any live transactional interface.
- Submission using HL7 SPL: The HL7 (Health Level 7) SPL (Structured Product Labeling) Protocol option allows the submission of GUDID device record data in XML format via FDA’s Electronic Submissions Gateway (ESG). To transmit via the ESG, you must register and complete ESG testing, and have a GUDID account set up (which also needs to be tested in advance). Using HL7 SPL for submission allows extensive pre-submission staging and testing prior to the final upload, and requires resources and training.
- Third-Party Submission: FDA will allow the use of a third-party submitter, i.e. a company or individual authorized to submit to the GUDID on behalf of the labeler. Third-party submission can be used in both the web-based and HL7 SPL submission approaches, and requires the labeler to designate the third party acting on their behalf. (For web interface submitters, labelers may designate a third-party as a coordinator or LDE user; for HL7 SPL submitters, labelers must identify third parties on their GUDID Account Request, and only those identified will be allowed to submit on behalf of labeler.) The third-party option allows organizations to engage experts experienced in GUDID data field assembly, formatting, error checking, and record transmission and save some time and money versus doing everything in-house.
Now That The Dust Has Settled, The Endpoints Are Clearer
The release of the UDI Final Rule was a relief for many — as limited direct part marking is required, multipacks need only bear UDI on the outside carton and even the alignment with the ISO 13485 date format, which is already in wide use.
But the clock is ticking and compliance dates are coming fast. If you need an extension for your Class III device, you will need to assemble your rationale before June 23, 2014, and be prepared to explain the compelling need for extension.
Also, assembly of GUDID elements cannot be an afterthought — it should be done in parallel with your UDI labeling workstreams, and all complex elements need to align. Consider outside professional help in assembly, formatting, error checking, and submission of GUDID data, and free up the necessary internal resources to focus on UDI labeling and manufacture issues. Finally, for Class II and Class I device manufactures, monitor the Class III submission process and try to glean lessons learned to help assembly of your records in the following years.
Author Bio: Tom Beatty leads the UDI Compliance Practice for QDevice, a division of QPharma, Inc. He has more than 18 years of life sciences market and project experience and is a graduate of the University of Notre Dame.
Acknowledgement: The author would like to thank Brendan Middleton of QPharma, Inc. for his editorial contributions.