
Bacterial Endotoxin Testing, Part 4: Key Tests For Endotoxin Detection
By Yadnyesh Patel, microbiology subject matter expert

With this article, I conclude my article series on bacterial endotoxin testing (BET). In my first article, I provided an overview of BET, including an introduction to the Limulus amebocyte lysate (LAL) method. In my second article, I discussed the preparatory requirements for the LAL test, reagent calibration/qualification, etc. In my third article, I discussed depyrogenation and calculating endotoxin limits and maximum valid dilution. In this article, I provide a deep dive on the following series of tests performed during BET analysis:
- labeled lysate sensitivity test
- non-interfering dilution determination (NID)
- validation for interfering factor
- test for bacterial endotoxin of samples/products.
Labeled Lysate Sensitivity Test
The lysate reagents received from vendors need to be verified for their labeled sensitivity. Confirm in four replicates the labeled sensitivity of λ expressed in EU/mL of the lysate prior to use in the test. The test for confirmation of lysate sensitivity is to be carried out when a new batch/consignment of lysate is received or used or when there is any change in the test conditions that may affect the outcome of the test. Prepare standard solutions having at least four concentrations equivalent to 2 λ, λ, 0.5 λ, and 0.25 λ by diluting the USP endotoxin reference standard (RS) with LAL reagent water. Mix a volume of the lysate with an equal volume (such as 0.1 mL aliquots) of one of the standard endotoxin solutions in each test tube. When single test vials or ampules containing lyophilized lysate are used, add solutions directly to the vial or ampule.
Perform the test on the four standard concentrations in quadruplicate and mark the tubes appropriately for negative control (NC) and control standard endotoxin (CSE) concentration. Add LAL reagent water (LRW), CSE, and lysate in each tube as shown in the table below.
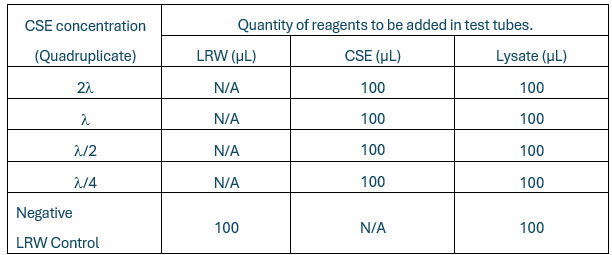
Table 1. “N/A” = Not applicable.
Incubate the reaction mixture for a constant period according to the directions of the lysate manufacturer (usually at 37 degrees C ± 1 degree for 60 ± 2 minutes), avoiding vibration. To test the integrity of the gel, take each tube in turn directly from the incubator and invert it through about 180 degrees in one smooth motion.
Interpretation Of Lysate Sensitivity Results
- If a firm gel has formed that remains in place upon inversion, record the result as positive.
- A result is negative if an intact gel is not formed.
- The test is considered valid when the lowest concentration of the standard solutions shows a negative result in all replicate tests.
What Are The Endpoint And Geometric Mean?
The endpoint is the smallest concentration in the series of decreasing concentrations of standard endotoxin that clots the lysate. Determine the geometric mean endpoint by calculating the mean of the logarithms of the endpoint concentrations of the four replicate series and then taking the antilogarithm of the mean value, as indicated in the following formula:
Geometric mean endpoint concentration = Antilog (Σe/f)
where:
Σe is the sum of the log endpoint concentrations of the dilution series used and
f is the number of replicate test tubes.
The geometric mean endpoint concentration is the measured sensitivity of the lysate (in EU/mL). If this is not less than 0.5 λ and not more than 2 λ, the labeled sensitivity is confirmed and is used in tests performed with this lysate.
Non-interfering Dilution Determination
The non-interfering dilution (NID) is the first set of positive product control (PPC) that shows a gel. Also, non-interfering dilution refers to the dilution that does not interfere with the test results. Screening for NID is performed prior to performing the validation for interfering factor for the BET.
Guidance for preparation of sample dilutions for NID test is described in Table 2:
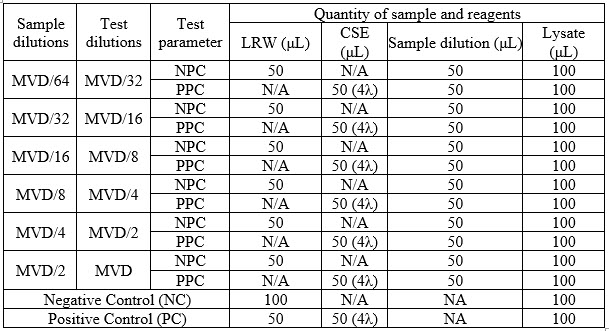
Table 2.
NC = Negative Control
PC = Positive Control<
NPC = Negative Product Control
PPC = Positive Product Control
N/A = Not applicable
After adding the sample, LRW, CSE, and lysate into the tubes, mix gently and incubate all the above tubes in the heating block for 60 minutes ± 2 minutes at 37 degrees C ± 1 degree C. After completion of the incubation period, observe all the incubated tubes by slowly inverting them approximately 180 degrees in one smooth motion.
Interpretation Of NID Test Results
- If a firm gel has formed that remains in place upon inversion, record the result as positive. (PC and PPC results should be positive.)
- A result is negative if an intact gel is not formed. (NC and NPC results should be negative.)
- NID should be less than the maximum valid dilution (MVD).
In cases where the NC, PC, and NPC results are within the acceptance criteria, but all the spiked dilutions of product (i.e., PPC) are found negative and if the sample under test does not comply with the test at a dilution less than the MVD, repeat the test using a greater dilution, not exceeding the MVD. The use of a more sensitive lysate permits a greater dilution of the sample to be examined, and this may contribute to the elimination of interference. Interference may be overcome by suitable treatment such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described above using the preparation to be examined to which standard endotoxin has been added and which has then been submitted to the chosen treatment.
Based on the NID results, MVD dilution shall be selected, and an interfering factor study shall be performed.
Based on NID test results, a recommendation can be made on the selection of product dilution for validation of the interfering factor. The selected non-interfering product dilution should be a minimum of one dilution more than the obtained endpoint or same dilution based on the feasibility of the product dilutions. This selected dilution is to be subject to the validation for interfering factor test in quadruplicate.
The NID test is the screening for the determination of the interference of product in the test; hence, it can be carried out on at least one batch of each product.
Validation For Interfering Factor On Bacterial Endotoxin Test (Inhibition/Enhancement Test)
To obtain accurate endotoxin measurements, inhibitors and enhancers must be identified and mitigated.
Based on NID test results, the recommended non-interfering product dilution of respective product is subjected to an inhibition/enhancement test in quadruplicate.
Although, historically, suitability tests using three consecutive lots of drug product were considered sufficient to assess suitability, it is recommended that an appropriate number of lots of product be determined prospectively for suitability testing to enable a valid assessment of the potential lot-to-lot variability in endotoxin content. This is especially important for biological products or products where product development and process validation have indicated significant lot-to-lot variability. Products with greater variability in their starting material, API, and manufacturing process will typically require more than three lots for suitability testing, whereas for products with little or no process or product variability, three lots may suffice. The number of lots tested for suitability should be supported by a risk assessment including information on the life cycle stage of the product (clinical/commercial), known sources of variability, and material testing history that could support an increased or decreased number of lots chosen for suitability testing. All materials that are being tested, including excipients and raw materials, should have a suitability study to ensure that any interferences and variability are identified, mitigated (if necessary), and taken into account in the testing procedures.
Common Test Interferences
Most pharmaceutical products have been found to interfere to some extent with BET performance. Because of the high assay sensitivity, these product-specific interferences can usually be overcome by dilution in water for BET not to exceed the product-specific MVD. Interferences may affect either the enzyme cascade of the LAL reaction itself or the analyte used as the PPC (e.g., purified lipopolysaccharide [LPS]), standard, or both.
The table below is a listing of common interferences and mitigations that may be considered and implemented beyond mere dilution. Where buffers or solvents other than water for BET are used for dilution, they should be free of detectable endotoxins.
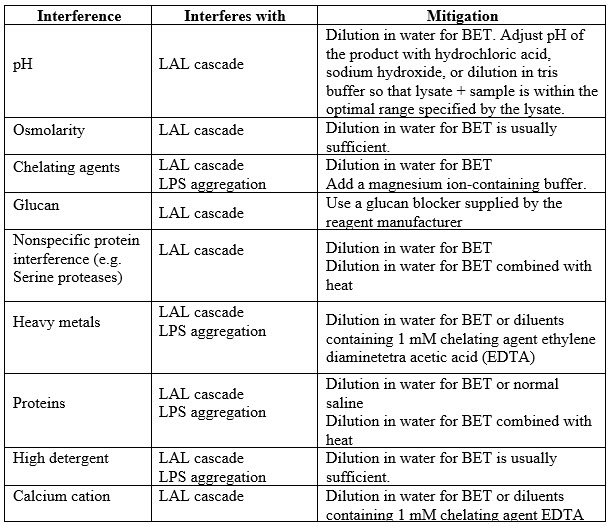
Table 3: Common Interferences and Mitigations
A majority of test interferences are overcome by simple dilution. Dilution can be in water for BET, buffers (e.g., tris or HEPES), buffers containing glucan-blocking agents, and buffers containing divalent cations or chelating agents as required. Dispersing agents may be added to diluents to further mitigate interference, and their value as an additive is typically considered during method development. It is noteworthy that glucan interference is a common interference in some biological products. Where glucan interference is likely, the use of a glucan blocker should be considered during assay suitability test method development.
Diluents other than water for BET and common buffers described above should be checked to assure that they do not interfere with the BET assay. For example, some neutralizing agents (e.g., proteinase K) may not be manufactured with appropriate controls against their contamination with GNB. If these reagents are used, they should be checked to assure that they do not contain endotoxins at levels that may contribute to the levels ultimately reported for the product. All BET assays must be conducted at neutral pH. Neutral pH for BET assays is generally defined as 6.0–8.0, but because all lysates are different in their formulations, the laboratory should confirm the proper range as described in the lysate manufacturer’s package insert. However, the reagent itself may provide buffering capacity so the mixture of lysate and product dilution should be tested for pH during the suitability study. pH may be adjusted to neutrality by using buffers as diluents or endotoxin-free hydrochloric acid (HCl) or sodium hydroxide (NaOH). If adjustment of pH is required using acid or base, it is suggested that the pH be measured during the suitability study to assess lot-to-lot variation in the product. Lysates from different manufacturers have different proprietary formulations, and all are calibrated for sensitivity against the RSE to ensure that they all detect LPS activity equally well in water. However, LPS activity may not be recovered equally in different products or by using different compendial test methods. If there is significant product-specific interference that cannot be overcome by dilution to within the MVD or by standard mitigation methodologies described in the above table, the laboratory should consider a different lysate formulation or test method before concluding that the LAL-bacterial endotoxins test is either invalid or unacceptable.
Preparation Of Sample Dilution
Sample dilution selected for validation for interfering factor on a bacterial endotoxin test should be less than MVD. Prepare the product/sample dilution according to the recommendation in the NID screening.
The example of sample dilution selection for validation is described below.
For a product where NID is identified as MVD/32 (i.e., recommended product dilution based on NID screening), then the selected test dilution should be a minimum of one higher dilution i.e., MVD/16. Prepare the sample dilution of MVD/32 and use for validation purpose.
Perform the test as per the following table.
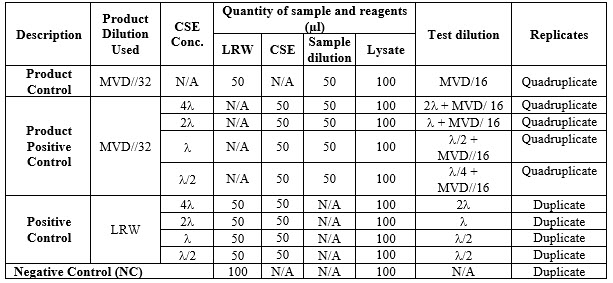
Table 4. “N/A” = Not applicable.
Reconstitute CSE and prepare a series of two-fold dilution of the CSE to give concentrations of 4 l, 2 l, l, and l/2. Reconstitute lysate as per manufacturer procedure.
Add the sample, LRW, and CSE in all tubes of PPC, NPC, PC, and NC as per the above table and then finally add 100 μL of lysate to all the tubes.
After addition of the sample, LRW, CSE, and lysate into all the tubes, mix gently and incubate all the tubes in the heating block for 60 minutes ± 2 minutes at 37 degrees C ± 1 degree C.
After completion of the incubation period, observe all the incubated tubes by slowly inverting them approximately 180 degrees in one smooth motion.
Find out the endpoint of concentration (geometric mean). The endpoint is the lowest concentration with the decreasing series of two-fold dilution of the control standard endotoxin (CSE) that shows a firm gel clot (positive result).
Calculate the geometric mean using the following formula:
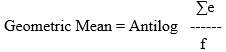
where:
∑e = Sum of Log10 endpoint concentrations of the dilution series
f = Number of replicate test tubes.
Calculate the average of the logarithm of the lowest endpoint concentration of endotoxin for which a positive reaction was observed. Take the antilogarithm of this mean. Then conclude the results of validation for interfering factors on the bacterial endotoxin test.
Acceptance Criteria
- Negative LRW Control (NC): Negative LRW control tubes should show negative results. No intact gel clot formation upon inversion of test tubes.
- Positive Control (PC): Positive control tubes should show positive results by firm gel clot formation, which remains in place upon inversion of test tubes.
- Negative Product Control (NPC): Negative product control (test sample) tubes should show negative results and no intact gel clot formation upon inversion of test tubes.
- Positive Product Control (PPC): Positive product control tubes should show positive results by firm gel clot formation, which remains in place upon inversion of test tubes.
The geometric mean value thus obtained in PPC and PC should be equal to or greater than 0.5 λ and equal to or less than 2.0 λ.
Interpretation Of Results Of Validation Test
If results of validation for interfering factors on BET are not found within the acceptance criteria, then the product test dilution is said to be “interfering with the test.” In that case, studies should be repeated considering following:
- Using a greater dilution that does not exceed the MVD.
- Using a more sensitive lysate permits a greater dilution of the sample to be examined.
- Interference of the sample solution or diluted sample solution may be eliminated by suitable treatment, such as filtration, neutralization, dialysis, or heat treatment.
- To establish that the treatment chosen effectively eliminates interference without loss of endotoxins, perform the assay described above using the preparation to be examined, to which standard endotoxin has been added and which has then been subjected to the chosen treatment.
- Endotoxin enhancement may be attributed to the presence of glucans in the samples and the presence of glucans may be overcome by using β-G buffers.
Revalidation for the test method is required when any changes are made to the experimental conditions that are likely to influence the test result.
Routine Testing For BET
Non-interfering dilution determination and method validation shall be performed of any product or sample prior to proceeding for routine BET. Such a method validation report shall be reviewed and approved by quality assurance personnel. Subsequent routine BET testing shall be performed by using the same dilution.
Precautions And Instructions
All materials used to sample materials for endotoxin content (scoops, pipettes, bottles) must be inert with respect to the material(s) being sampled, and they must be sterile and free of detectable endotoxins. Even though samples are often taken in the field without the aid of a laminar flow hood, samplers must take precautions that
a) they do not contaminate the sample itself, and
b) in taking the sample, they do not contaminate the rest of the material in its original container.
Historically, the sampling scheme for finished drug products is to take at least three units representing the beginning, middle, and end of the batch. However, for a standard run of a small-volume parenteral, biological therapy, or large-volume parenteral, three units may not be a representative sample and may identify only those lots that are uniformly and highly contaminated. Sampling schemes should be justified and should be based on the known variability of the process, the unit operations of the process, historical knowledge of the process, and materials used in manufacture.
Bacteria and endotoxins are generally not homogeneously distributed in any product or material, liquid, or powder. Care must be taken to flush ports in a circulating water loop before sampling and use the same equipment for sampling that is used for manufacture to assure a representative sample. For example, if a hose is attached to a port for the purpose of transferring water from a circulating loop to another formulation vessel, the water sample must be taken from the same hose with the same flush times as used in manufacturing. So, while the water within a properly constructed and controlled loop may theoretically be homogeneous, differences in the ports along the distribution loop may add endotoxins to samples. For highly viscous materials, a suitable inert rod free of detectable endotoxins may be used to mix the material prior to sampling. Sterile, pyrogen-free spatulas and scoops are used for powdered and granular solids. Because of the lack of assurance of homogeneity, the number of samples from viscous materials and powders should be determined relative to the number of units initially received using appropriate statistical procedures. Particularly for samples of viscous and powdered materials, samplers should be trained on how to spot signs of non-uniformity. Such indicators may include differences in shape, size, or color, or evidence of moisture in powdered materials. For viscous materials, samplers should be trained to look for stratification of the material, differences in viscosity or colors, and particulate contamination. Samples of raw materials that could exhibit variability in endotoxin content should not be pooled.
In the case of a new vendor of a raw material, the methodology and calculations used to determine the endotoxin content of the material should be reviewed. If the material is designated as critical for manufacturing, a site audit of the vendor may be in order. In addition, it is strongly suggested that an in-house confirmation of the accuracy of the endotoxin level stated on the COA be performed. For critical materials, the quality agreement should require that manufacturers of these materials inform the pharmaceutical manufacturer of changes in processes, controls, or raw materials so that the changes can be discussed at change control and the need for re-confirmation of materials can be determined. Once the COAs have been successfully confirmed for a predetermined number of shipments justified in the sampling plan by history, and depending on the variability of the manufacturing process, the laboratory can consider accepting the COA with periodic testing of the incoming materials, as long as the manufacture of the materials has not changed.
BET Testing Can Be Performed On Pooled Samples
The term "pooling" refers to creating a composite sample preparation that includes the total contents of several individual units or equal aliquots from the units taken from the same lot or batch. Pooling is often done for batch release testing. Pooling is an acceptable option for laboratories performing any of the BET assays on drug products. However, pooling has a number of drawbacks; the most obvious is that it will dilute endotoxins that may be in any one of the units, potentially concealing the variability in endotoxin content and distribution among the units that were pooled. Sampling plans, including instructions to pool samples, should be scientifically justified. If units are pooled, the MVD must be adjusted to account for the possibility of endotoxins in just one of the samples. The adjusted MVD is calculated by dividing the originally calculated MVD by the number of units contributing to the pool. For example, if the MVD for a small volume parenteral is 240 and a manufacturer chooses to pool three vials for testing, the adjusted. MVD is 240/3, which is 80. This reduction in MVD effectively reduces the endotoxin limit for the product by a factor of three to compensate for pooling. While pooling may result in an incremental savings in reagent usage and therefore save some money, there are a number of points to consider when pooling:
- Pooling may obscure any non-uniformity in endotoxin content between the individual sample units. Information on variability may be valuable in troubleshooting or investigations. For example, random contamination in one of multiple filling needles may cause some vials to contain endotoxins and others not.
- Taking aliquots of samples for pooling should always be performed using aseptic technique and with individual units vigorously mixed prior to removing the aliquots. The original containers with remaining product should be retained for investigation in the event of an out of specification (OOS) test result. Removing the aliquot through the disinfected rubber stopper using a pyrogen-free syringe is advisable to maintain the integrity of the unit container during subsequent storage for investigative testing.
- The concept of adjusted MVD does not apply to medical devices as they are, by convention, commonly pooled for testing.
- Any sampling scheme for drug products must represent the beginning, middle, and end of the batch. Additional samples may be taken if interventions in manufacturing raise concerns about possible endotoxin contamination.
- If testing at the adjusted MVD causes an unacceptable increase in product-specific interference, samples should be tested individually.
- Products with low calculated MVDs, or suspensions where there is no assurance of homogeneity in the removed aliquots, may not be good candidates for pooling.
- Pooling is not appropriate for in-process samples, particularly those representing different stages of manufacturing.
Bacterial endotoxin testing is one of the significant tests considered as release parameters and batch release decision makers in pharmaceutical industries.
These Guidelines Will Help You Throughout The BET Process:
- USP chapter <85> Bacterial Endotoxins Test
- USP chapter <1085> Guidelines On The Endotoxins Test
- ICH guideline Q4B
- General Test 4.01 Bacterial Endotoxins Test (Japanese Pharmacopoeia)
- General notices 2.6.14: Bacterial Endotoxin Test European Pharmacopoeia (Ph. Eur.)
- General notices 2.6.32: Test for bacterial endotoxins using recombinant factor C (Ph. Eur.)
About The Author:
 Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, at Zydus group, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.
Yadnyesh Patel earned his master’s degree in microbiology from KBCNM University, Maharashtra, India, in 2009. With over 13 years of extensive experience in quality functions, he has spent time working at pharmaceutical organizations such as Claris Otsuka Ltd, Sun Pharmaceutical Industries Ltd., and Zydus Life Sciences Ltd. Currently, at Zydus group, he spearheads microbiological quality functions, document management, audit, and compliance. He possesses substantial expertise in quality management systems, SOPs, documentation management, microbiological test method validations, sterility assurance, aseptic process simulation, and computer system validation.