
Brexit Finalization: How Will Medical Device Compliance Change In Europe In Jan. 2021?

The UK left the European Union (EU) earlier this year with an 11-month transition period ending on December 31, 2020. Given that the UK has been a member of the European Union for almost 46 years, the bilateral interactions are deeply rooted and the mechanisms to ensure hassle-free interactions between the UK and the EU after the exit is crucial. After Brexit's transition period, high import tariffs, duties, and taxes would increase the cost of the device, making them less competitive in both the UK and the EU. The Medicines and Healthcare products Regulatory Agency (MHRA), which regulates medical devices marketed in the UK, released new guidelines in September 2020 detailing how devices will be regulated beginning January 1, 2021, with varied compliance dates for different device classes. The new rules will be applicable for placing medical devices in Great Britain, Northern Ireland, and the EU. Medical device and in vitro diagnostic (IVD) manufacturers will need to register medical devices with the MHRA, identify a UK Responsible Person, obtain proper device certification, and include proper device marks and labels.
The location of the manufacturer, the risk classification of the device, and the market of interest determine the regulatory requirements for device manufacturers to place a new device into the market and for continuity of existing device distribution. The enforcement powers of the MHRA are derived from the four main regulations noted below.
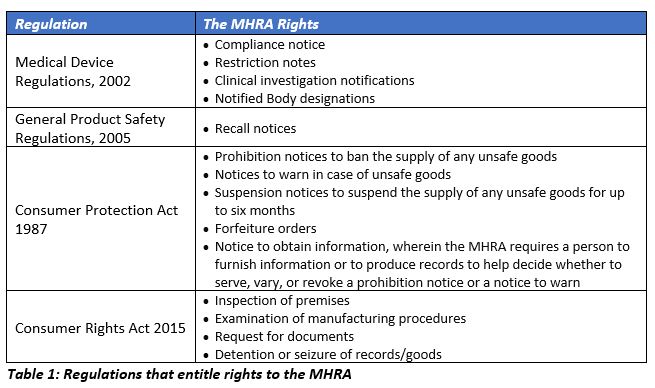
The Medicines and Medical Devices Bill is currently under review by the House of Lords and would be in effect after the Royal Assent. To support the medical device industry after the transition period, more regulations and guidelines are expected to be released from time to time. Until the end of transition period on December 31, 2020, the laws prevailing in the EU, as well as the single market and customs union, are in effect in the UK, as per the 2002 Medical Device Regulations (MDR). The UK cannot be a part of the EU decision-making group.
Prior to Brexit, device manufacturers in the UK are currently complying with Directive 90/385/EEC on active implantable medical devices (EU AIMDD), Directive 93/42/EEC on medical devices (EU MDD) and Directive 98/79/EC on IVD medical devices (EU IVDD) through the end of 2020 in order to place devices in the UK market. The MHRA regulations also required the manufacturers of all class I devices manufactured, refurbished, or re-labelled with manufacturer name; any system or procedure pack containing at least one medical device; and custom-made devices and IVDs manufactured and/or undergoing performance evaluation to register with the Health Authority. All other devices falling into any other device class are required to have the conformity assessment carried out by the Notified Bodies (NBs) located in any of the EU countries (known as the EU 28 before Brexit, now the EU 27) in order to place the device in the market.
The geographies are grouped as Great Britain, Northern Ireland, and the EU 27 to assess the impact of Brexit on the medical device industry and to create an action plan for various stakeholders. The geopolitical scenarios in Northern Ireland require the regulations to be different from those to be implemented in Great Britain.
Device Registration with the MHRA
As of January 1, 2021, all devices, irrespective of device class, must be registered with the MHRA. This enforcement has a grace period of varying duration, based on risk class of the device.
- High-risk Class III and Class IIb implantable devices must be registered within four months of the date of enforcement.
- Moderate-risk Class IIa and other Class IIb devices have a grace period of eight months.
- Low-risk Class I devices have varied implementation timelines in the UK and Northern Ireland. These devices are required to be registered immediately from January 1, 2021, to be marketed in Northern Ireland, whereas a grace period of 12 months is available for devices to be marketed in the UK. This grace period in the UK does not apply to the Class I, IVD, and custom-made devices that required registration with the MHRA even before the decision to exit the EU was taken.

Figure 1: Post-Brexit Compliance Timelines for varied device classes in Great Britain and Northern Ireland
Identifying and Appointing a UK Responsible Person
Non-UK medical device manufacturers need to appoint a UK Responsible Person (UKRP), who must register with the MHRA. The manufacturer shall have a registered place of business in the UK to register the devices with the MHRA and to take responsibility of the device being placed in the UK. Importers or distributors representing the manufacturers can act as UKRPs and can liaise with the MHRA. As detailed in the UK MDR 2002, the roles and responsibilities of the UKRP are:
- To ensure that the technical documentation is available and updated in a timely manner
- To ensure the conformity assessment of the device is successfully concluded
- To ensure the Declaration of Conformity (DoC) is in place
- To have all the technical information about the device and to produce the details and device samples whenever requested by the MHRA. In case of non-availability of samples, the UKRP may coordinate with the manufacturer and ensure that the samples are submitted.
The UKRP will also be playing a critical role in the post-market surveillance of the device and should work with the manufacturer and the MHRA to implement corrective action and preventive action associated with complaints and related safety issues.
The European Authorized Representatives (EARs) based out of the UK will no longer be recognized by the EU and the manufacturers shall appoint a new EAR from one of the EU 27 countries or Northern Ireland. Manufacturers from countries other than the UK and the EU need to appoint an EAR (with roles and responsibilities defined in the MDR and IVDR) to market the device in Northern Ireland.
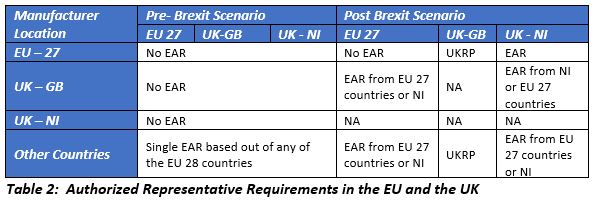
Changes to NBs and CE Certifications
Prior to Brexit, owing to the single market concept, CE certification issued by any of the NBs based out of any EU country was recognized across the UK and the EU. After Brexit, manufacturers intending to place the device or market the device in the EU 27 need the CE certificate issued by the NB based in the EU 27. The MHRA continues to recognize CE marking and the certificates issued by the NBs located in the European Economic Area (EEA) until June 30, 2023. Devices certified by the UK NB should transfer its certificates to the NBs based out of the EU 27 in order to continue marketing in the EU. The majority of the NBs based in the UK are partnering with NBs based in the EU to facilitate smooth transfer of certifications.

Changes to Marks and Labels
By July 1, 2023, all devices marketed in Great Britain need to have UKCA marking. In Northern Ireland, the UKCA mark is not valid and the manufacturer may opt for either the CE mark (as per IVDR) or the CE UK(NI) mark.
The UK-based NBs approved under the MDD, IVDD, and AIMDD will be automatically designated as Conformity Assessment Bodies (CABs) for the UKCA marking in effect January 1, 2021. Beginning on that date, the MHRA will initiate designating the CABs. Manufacturers intending to market devices in Northern Ireland can get the conformity assessments from the NBs based out of either the UK or the EU 27. In Ireland, the marking on the label would depend on the location of the NB.
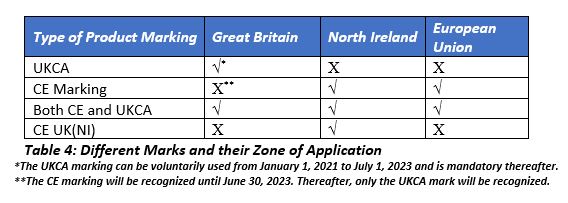
The devices to be placed and marketed in the EU 27 and Northern Ireland must comply with the EU labeling requirements. Unlike the pre-Brexit market, in which a single CE marking was required, post-Brexit markets would have three markings (one for Great Britain, one for Northern Ireland, and one for the EU). The devices intended to be placed in Northern Ireland should have the CE(UK)NI mark (if using a UK-based CAB) or the CE mark (if using an EU-based NB).
Going Forward: 2021 and Beyond
Unlike in Great Britain, the EU MDR and EU IVDR will be implemented in Northern Ireland starting in May 2021 and May 2022, respectively, along with the other EU 27 countries. Distributors of UK-manufactured devices will be considered as importers in the European Union and would be subject to product liability requirements of the device, as per Product Liability Directive 85/374/EEC. The detailed regulatory requirements in each geographic area are summarized in the below table.
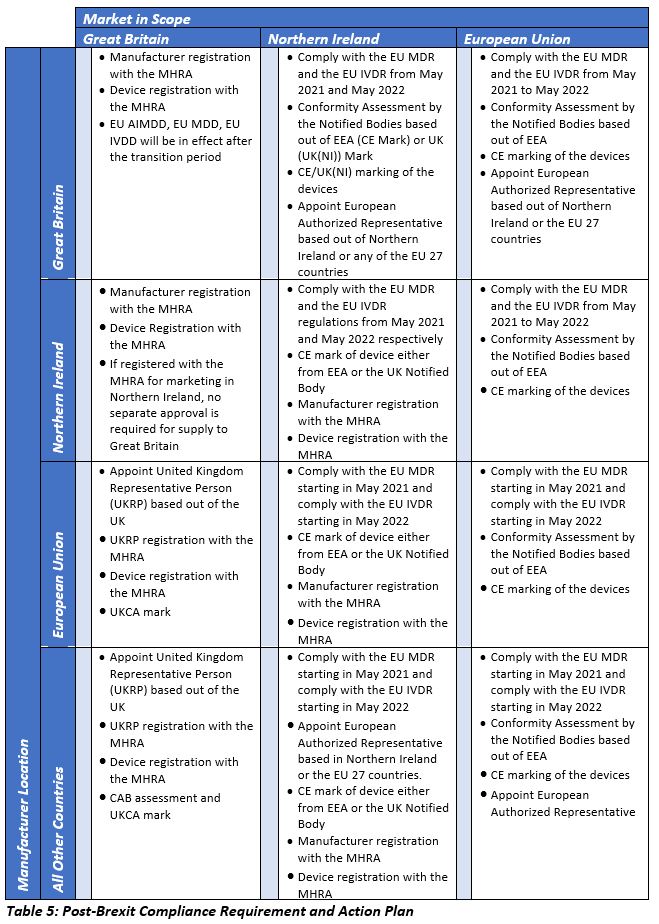
Non-UK device manufacturers willing to launch their devices for the first time in Great Britain should comply with the EU MDD requirements and should initiate registration of Class I devices or perform conformity assessments for devices of other classes. Manufacturers with devices already being marketed in Great Britain can continue marketing their Class I devices, provided that the devices are already registered with the MHRA. Manufacturers of other device classes should initiate the discussion with their current NBs for any collaboration with their counterparts in the UK. If not, the manufacturers should find the UK-based CABs for recertification. In all the above scenarios, the first step is to identify and appoint a UKRP followed by the other activities as mentioned above.
About The Authors
 Induja Ratakonda is a solutioning and pre-sales lead at Freyr, a global regulatory service provider. She has a degree in pharmaceutical marketing and has comprehensive understanding of end-to-end product lifecycle management. She has handled complex multi-year projects that range from initial development to regulatory submissions and commercialization. She has keen interest analyzing the global regulatory landscape and the impact of regulatory dynamics on the manufacturing industry.
Induja Ratakonda is a solutioning and pre-sales lead at Freyr, a global regulatory service provider. She has a degree in pharmaceutical marketing and has comprehensive understanding of end-to-end product lifecycle management. She has handled complex multi-year projects that range from initial development to regulatory submissions and commercialization. She has keen interest analyzing the global regulatory landscape and the impact of regulatory dynamics on the manufacturing industry.
 Vardhini S. Kirthivas has more than 20 years of experience and leads Strategic Services at Freyr, including the Medical Devices Center of Excellence. She is responsible for solutioning and project delivery of global medical devices and in vitro diagnostic devices. She has led projects related to niche medical devices involving latest technologies such as software and AI as well as complex regulatory submissions in the U.S., Canada, EU, Latin America, and Asia-Pacific markets. She worked for many multinational companies before joining Freyr.
Vardhini S. Kirthivas has more than 20 years of experience and leads Strategic Services at Freyr, including the Medical Devices Center of Excellence. She is responsible for solutioning and project delivery of global medical devices and in vitro diagnostic devices. She has led projects related to niche medical devices involving latest technologies such as software and AI as well as complex regulatory submissions in the U.S., Canada, EU, Latin America, and Asia-Pacific markets. She worked for many multinational companies before joining Freyr.