
Building The AI-Enabled Medical Device QMS For European Compliance
By Srividya Narayanan, MSc, CQSP, Asahi Intecc; and Apekshit Mhatre, Northeastern University

With the increasing use of AI in medical devices, there are complex requirements, as ISO 13485:2016 serves as the foundation for quality assurance in medical devices.1 We now have a quality regulation in place, as outlined in the EU AI Act, with Article 17 providing specific requirements for AI.2 Internationally, we had a gap filled with ISO/IEC 42001:2023, which provides the first standard for AI management systems.3
Within the EU's regulations, Article 10(9) refers to quality system requirements under Annex IX. This is a specific requirement concerning quality control for the devices that use AI technology under their design or functionality.4 This is where the complexity begins. It creates an important question in terms of strategic planning on the part of manufacturers: Do we need to develop a quality system in order to handle each framework individually, or can we merge them together into one efficient system? It directly affects pricing and competitiveness in both the U.S. and EU markets.
Regulatory Authorization For QMS Integration
The EU AI Act particularly understands and promotes quality management system integration. Article 17(2) of the Artificial Intelligence Act provides that for providers of high-risk AI systems that are subject to quality management requirements under sectoral union law, "the aspects described may be part of the quality management systems pursuant to that law." This provision allows medical device manufacturers to integrate requirements from Article 17 of the Artificial Intelligence Act into their existing quality management systems based on ISO 13485 rather than building a system management infrastructure for AI.5
This was strengthened by the Medical Device Coordination Group 2025-6, which clarified that the obligations for quality management systems under the AI Act “may be integrated” with MDR quality management systems to achieve complementarity and reduce administrative burden.6 Those manufacturers that continue with ISO 13485 should integrate Article 17 obligations into current quality management processes, rather than using the stand-alone standard ISO/IEC 42001.
The effect of this integration requirement has more practical applications than mere convenience for regulatory purposes. The responsibility for performing a single compliance assessment for the combined requirements has the potential to be carried out by the notified bodies for the MDR and AI Act frameworks.7 This, however, requires careful planning that does not give less priority to the requirements of either framework.
Four-Standard Foundation
Before designing a comprehensive system, it is necessary to understand the unique emphasis and role of each standard – ISO 13385:2016; ISO/IEC 42001:2023; MDR Article 10(9) + Annex IX; and Article 17.
Standard 1: ISO 13485:2016
ISO 13485:2016 specifies QMS requirements for organizations involved in the design, development, and delivery of medical devices with quality and safety throughout the product life cycle.8 Medical device design controls (Section 7.3) require systematic planning, input identification, output documentation, design review, verification, and validation procedures.9 Manufacturers obtain ISO 13485 certification through a separate third-party agreement, ensuring quality systems comply with internationally accepted standards for quality, consistency, and effectiveness. Although MDR Article 10(9) does not require ISO 13485 certification, the match between MDR Article 10(9) and ISO 13485 makes certification a necessity in industry, and buyers demand evidence of conformity.
Standard 2: ISO/IEC 42001:2023
ISO/IEC 42001:2023 establishes, implements, maintains, and constantly improves AI management systems. In December 2023, it was announced that the initiative is in line with the U.S. Risk Management Framework and the EU AI Act.10 This shows how important AI is becoming in running a business. Requirements include defining organizational context, management commitment and involvement, managing risks and opportunities, competence and awareness, and operational controls. ISO/IEC 42001 follows a process-oriented approach designed for integration with other management standards such as ISO 9001, ISO 13485, and ISO 27001, enabling organizations to demonstrate responsible AI development through improved controls on quality, security, and transparency.3 ISO 42001 certification is not required, though. This is because Article 17 of the AI Act can be met by extending ISO 13485 systems without having to implement ISO 42001 separately.
Standard 3: MDR Article 10(9) + Annex IX
MDR Article 10(9) and Annex IX outline the responsibilities of a quality management system (QMS) for medical devices.4 These include planning for regulatory compliance, making sure that the device meets safety and performance standards, allocating management responsibilities and resources effectively, planning for risk management, providing evidence of clinical evaluation, planning for post-market surveillance, communicating with authorities, managing vigilance and corrective/preventive actions, and analyzing data for continuous improvement.11 Annex IX shows the most common compliance process under MDR, which is used most often by manufacturers that follow ISO 13485. Applications filed with notified bodies provide detailed overviews of QMS conformity, and successful certification enables EU market access.12
Standard 4: AI Act – Article 17
The AI Act requires that QMS for high-risk AI systems ensure regulatory compliance, documented in written policies, procedures, and guidelines covering 13 aspects, including regulatory compliance strategies, design labeling, and staff resource management responsibilities. The data management requirements (Article 17.1(f)) are highly relevant to AI medical devices, requiring documented procedures covering all stages of data management from acquisition to long-term storage.13 This extends beyond usual medical device documentation to address AI-specific considerations, including data set representativeness, training, and validation requirements.
Building The Integrated System: Five Key Components
The five-part framework shall be cohesive, building from ISO 13485 foundations to cover the AI Act Article 17 requirements while drawing upon relevant best practices found within ISO 42001. A unified system means compliance is ensured in one place rather than having to manage different tracks.
Component 1: Strengthened Management Supervision and AI Governance
A traditional device quality management system is primarily focused on manufacturing and post-market surveillance. Adding AI governance introduces concepts such as fairness in algorithms, data governance, transparency, human oversight, and ethics. Create a cross-functional AI governance committee that includes representatives from clinical, data science, compliance, quality, cybersecurity, ethics, and patient advocates. This committee reviews AI projects, formulates AI-related policies, aligns AI interests with regulatory obligations, and attends to ongoing life cycle monitoring.14
Establish an integrated AI policy file (as required in Article 17.1(a) of the AI Act), which harnesses and enhances current policies of quality. Outline your organizational strategy and guidelines for responsible AI use. Define your organization’s ethics for fairness and transparency. Outline your strategies and processes for compliance with MDR and AI Act regulations. Outline your process for updating algorithms. Provide your organizational investment in AI-related human resources and infrastructure. 15
Component 2: Embed AI-Specific Risk Management
Envision the extension of the application of ISO 14971:2019 risk assessment criteria to the domain of AI and consider the following risks associated with algorithms and the application of those algorithms in the medical domain. These risks might include the following. Implement risk scoring: Risk scores of 70-100 require immediate mitigation, scores of 40-69 mandate frequent observation, and scores of 1-39 necessitate review. Risk scores should have designated owners.16
List the AI-specific factors found in the integrated risk file: training data characteristics, validation constraints, ways in which performance could differ for different groups, intended use environments, ways in which failure of human oversight could occur, cybersecurity risks, and drift detection systems.17
Component 3: Comprehensive Data Governance Infrastructure
Data governance is a paradigm shift of unprecedented proportion. AI necessitates end-to-end data set governance, whereas traditional device QMS concentrates on manufactured data. Before beginning AI projects, establish standard operating procedures regarding:
- Guidelines for obtaining the data sets as well as the quality standards expected by you
- Procedures for data cleansing with strong traceability
- Qualifications of annotators and inter-rater reliability metrics
- A clear division between training and test sets, that is, test data should not be used in training 18
- Ensuring GDPR/HIPAA compliance19
- Versioning and change management
- Impact assessment reports regarding any modifications to data sets
Conduct routine audits of the demographic distribution, including age, gender, race, ethnicity, socioeconomic status, geography, and comorbidities.20 Determine how underrepresented groups require focused data collection efforts by comparing these with the target audiences.
Component 4: Algorithmic Transparency and Human Oversight Mechanisms
Articles 13 and 14 of the AI Act aim to encourage transparency and human control in addressing the so-called “black box” problem in machine learning. The algorithm's architecture, the features of the input variables, and how they relate to medical practice must be explained in the technical documentation needed for transparency. The model's limitations and contraindications must also be clearly stated.21
Human oversight, under Article 14, is mandatory. This includes real-time monitoring displays of confidence levels and quality of results, pause, override, or halt functions for the system, audit trails for recording the results of the system and user choices, and alerting functions for unusual inputs and low-confidence results.21 These should be verified through usability testing with representative clinicians for their ability to judge when to make an intervention and to carry out an override.
Component 5: Automated Logging and Post-Market Performance Monitoring
Under the AI Act, Article 19, there is a mandatory logging requirement regarding post-market monitoring. This refers to continuous, automatic data collection, which should not depend on intermittent human observations. Logging should record all aspects and needs to be secure and tamperproof. This requires its storage for the lifespan of the medical device and additional periods ranging from 10 to 15 years.
Performance drift detection is a necessary step because ML algorithms may drift as a reflection of changes occurring in the manner of practice, the demographics of patients, or the data inputs.22 Performance needs to be defined, a baseline established, and statistical process control implemented to identify changes; triggers should then be defined for further investigation and appropriate interventions of retraining or withdrawal. 23 Develop dashboards to monitor algorithm activity in real time on all operational sites and integrate these monitoring systems with the MDR surveillance work in the industry, allowing data to flow into periodic safety update reports and post-market clinical follow-up studies.24
Bias Mitigation Across The Product Life Cycle
The AI Act states that biases should be detected and corrected at all stages of a product’s life cycle.25 Although safety is a key element in the standard ISO 13485 and the MDR, it neither specifies nor regulates bias control.
- Conception: To come up with solutions, assemble a well-rounded team of clinicians, data scientists, and advocates for health equity. In addition, include patients from underrepresented groups to share their insights.17
- Data collection: Focus on the ability of the data to represent the real world for age, gender, racial, ethnic, socioeconomic, geographic, and existing health condition variables.25 If this isn't feasible, note the areas of deficiency, the clinical implications, and any relevant warnings or contraindications.
- Building a model: Incorporate fairness strategies such as optimization with fairness constraints, impact through reweighting for underrepresented subgroups, balance through resampling, and annotation bias correction through relabeling.26,27
- Validation: Extract and decompose model evaluation results into accuracy, sensitivity, specificity, positive predictive value, and negative predictive value and test for differences among demographic subgroups.20
- Post Deployment: Implement real-world performance monitoring for demographics in real-world settings. In cases where a demographic has underperformed, take corrective action: Retrain models, change model uses, improve notifications, and, as an extreme action, leave markets.28
Implementation Road Map
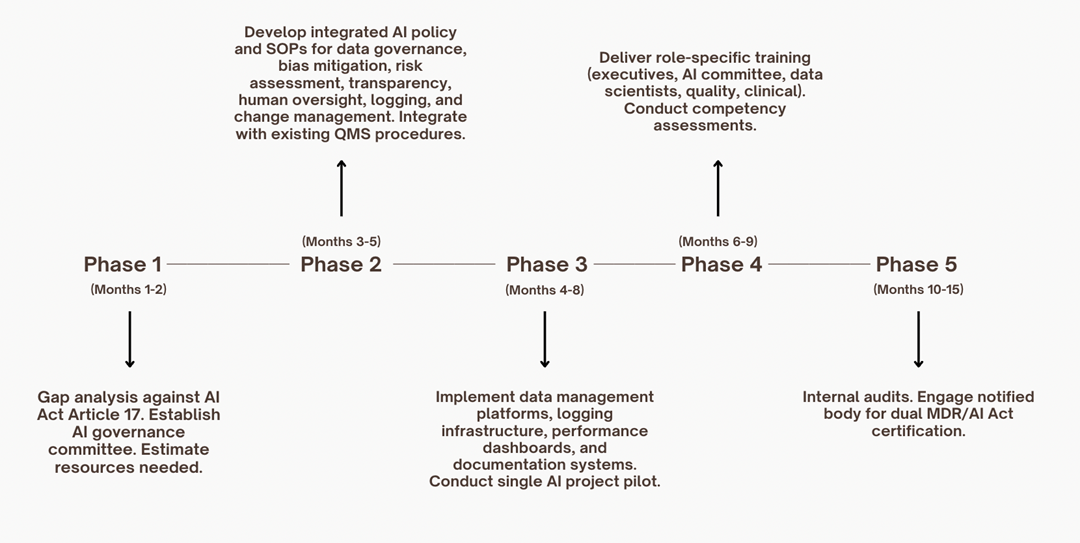
For more information on these phases, see references 14, 18, 23, 29, and 30.
Common Pitfalls And Solutions
Pitfall 1: Developing Disjointed, Non-Integrated AI Systems
Pitfall: Businesses create separate AI quality teams that operate concurrently with current quality departments. The result is misalignment, careless documentation, and resource waste. To put it briefly, parallel tracks result in duplicative work and communication gaps.14
Solution: Develop and expand existing quality teams rather than spawning new ones. Place AI governance in the hands of the existing quality management representatives. Incorporate the requirements for AI into existing procedure templates, extending risk management to include AI-specific hazards rather than creating new documents. Perform combined audits so that notified bodies can assess the MDR and AI Act compliance at the same time.30
Pitfall 2: Poor Data Governance Foundation
Pitfall: Traditional devices and workflows don't require full tracking of a data set's life cycle. AI projects fail to advance because developers are unable to trace or validate the data segregation. Too often, teams find out too late that hidden biases exist in the data sets or that the training and test data have been mixed, rendering performance claims dubious.18
Solution: Make upfront investments in data management systems. As a prerequisite for AI projects, establish data governance procedures and the underlying technology. Create systems with automated data set separation, audit trails, access controls, and version control. Before approving any AI development work, a formal data governance plan should be required.18
Pitfall 3: Bias Mitigation
Pitfall: Identify the issue toward the end, after final testing, and you are left with limited and expensive ways to remedy it. This presents a dilemma for the manufacturer. Do they shrink the desired application, collect new data, and retrain the model (which will now delay the project) or simply abandon the project altogether, which will cost profoundly?27
Solution: Incorporate bias observation from inception to completion of models, using mixed groups comprising people from representative subgroups of recipients.20 Perform repeated bias assessments as you source data to create models. Establish a bias threshold to correct models before validation; a margin of more than 5% in differences in performance of models across subgroups necessitates examination before proceeding.26
Pitfall 4: Lack of Transparency for Clinical User
Pitfall: Teams are capable of achieving a high level of accuracy but refuse to describe how their algorithms work or when their output is valid. This could mean physicians are adhering to incorrect recommendations blindly or ignoring correct recommendations, which would be a risk to patient and manufacturer liability issues because of a violation of trust.21
Solution: Perform usability studies on the service with target clinical users during the development process. It should be ensured that the transparency mechanism facilitates appropriate trust calibration. Scenarios in which the limitations of the algorithm prevent clinical use should be recorded. The intervention process should be tested under realistic conditions after considering the effects of time pressure and cognitive load.21
Pitfall 5: Post-market Monitoring
Pitfall: Huge investments are being made for premarket validation, and there is a lag in monitoring once the product enters the real world.22 The consequences are that performance drifts and biases creep in unnoticed. Holding onto the assumption that premarket performance will translate into future accuracy neglects the fact that clinical practices evolve and the demographics of patients influence the erosion of the behavior of the algorithm. Patients can be harmed before problems are noticed.28
Solution: Implement automated logging and monitoring from the start, before launching the tool or service. This includes specifying thresholds that would trigger concern, from either a decrease of more than 3% from the validation baseline or a demographic subgroup falling below the satisfactory performance level. The quarterly monitoring would measure performance against validation baselines, budgeting post-market funds proportionate to the cost of the qualitative investment.28
What To Expect In The Future?
The CEN/CENELEC Joint Technical Committee 21 is busy working on the harmonized AI Act standards. This could result in proposed changes as early as 2026-2027.31 Now the FDA's requirement of ISO 13485:2016, which takes effect in February 2026, moves the U.S. and the EU a step closer.32 This could prove to be a key milestone because Subtle Medical has attained ISO 42001:2023.33
For the manufacturing sector, an investment approach to integration is beneficial. This is because it enables faster algorithm development through the use of common data sets, makes the technology more appealing and reduces the liability risks associated with bias, offers quick solutions to any problems through monitoring, and makes interaction with regulatory bodies easier. Effective quality management is then an area of competitive advantage.34
An integrated QMS framework and approach provide the answer, combining various regulations into one efficient way of working. Organizations embracing this method are not only meeting regulations, but they are also demonstrating operational excellence, which supports innovation, quality, and patient safety throughout the entire life cycle of their artificial intelligence medical device.
References
- ISO 13485 | Quality Management Systems for Medical Devices. Intertek. March 31, 2025.
https://www.intertek.com/assurance/iso-13485/ - Article 17: Quality Management System | EU Artificial Intelligence Act. Artificial Intelligence Act EU. November 30, 2023.
https://artificialintelligenceact.eu/article/17/ - ISO/IEC 42001: AI management systems. Johner Institute. March 2, 2025. https://blog.johner-institute.com/quality-management-iso-13485/iso-iec-42001/
- MDR - Article 10 - General obligations of manufacturers. Medical Device Regulation EU. July 3, 2019.
https://www.medical-device-regulation.eu/mdr-article-10-general-obligations-of-manufacturers/ - AI Act: MDR Guidelines for Medical Device Manufacturers. QuickBird Medical. January 12, 2026.
https://quickbirdmedical.com/en/ai-act-medizinprodukt-mdr/ - EU Guidance MDCG 2025-6 Clarifies Compliance for Medical Device AI. Pure Global. July 21, 2025.
https://www.pureglobal.com/news/eu-guidance-mdcg-2025-6-clarifies-compliance-for-medical-device-ai - Digital Omnibus and AI Act updates Key implications for MedTech. DLA Piper. November 30, 2025.
https://www.dlapiper.com/en/insights/blogs/cortex-life-sciences-insights/2025/digital-omnibus-and-ai-act-updates-key-implication - AI Device Standards You Must Know - ISO 13485, 14971, 62304. Hardian Health. March 12, 2025.
https://www.hardianhealth.com/insights/regulatory-ai-medical-device-standards - ISO 13485:2016 7.3: Medical device design controls and why they're important. Ideagen. June 11, 2025.
https://www.ideagen.com/thought-leadership/blog/iso-134852016-73-medical-device-design-controls-and-why-they-re-important - ISO/IEC 42001: Artificial Intelligence Management Systems (AIMS). ANSI. May 20, 2025.
https://blog.ansi.org/anab/iso-iec-42001-ai-management-systems/ - EU MDR Quality Management System (QMS). SimplerQMS. June 16, 2025.
https://simplerqms.com/eu-mdr-quality-management-system/ - Understanding MDR Annex IX. Conformity assessment. MDRC. December 31, 2024.
https://mdrc-services.com/mdr-annex-ix/ - AI Act Service Desk - Article 17: Quality management system. European Commission. June 12, 2024.
https://ai-act-service-desk.ec.europa.eu/en/ai-act/article-17 - 6 steps to successful ISO 42001 implementation. CIS-Cert. June 16, 2025.
https://www.cis-cert.com/en/news/6-steps-to-successful-iso-42001-implementation/ - Healthcare AI Data Governance: Privacy, Security, and Vendor Management. Censinet. January 31, 2025.
https://censinet.com/perspectives/healthcare-ai-data-governance-privacy-security-and-vendor-management-best-practices - ISO 42001: Ultimate Implementation Guide 2025. ISMS.online. September 17, 2025.
https://www.isms.online/iso-42001/iso-42001-implementation-a-step-by-step-guide-2025/ - Bias recognition and mitigation strategies in artificial intelligence healthcare. Nature. March 10, 2025.
https://www.nature.com/articles/s41746-025-01717-9 - AI Data Management & Governance: Best Practices for Compliance. Rephine. February 27, 2025.
https://www.rephine.com/resources/blog/ai-data-management-governance-best-practices-for-compliance/ - Cybersecurity requirements for medical devices in the EU and US. PMC. July 14, 2025.
https://pmc.ncbi.nlm.nih.gov/articles/PMC12301760/ - Mitigating Bias in AI-Enabled Medical Devices. Clarkston Consulting. July 24, 2023.
https://clarkstonconsulting.com/insights/mitigating-bias-in-ai-enabled-medical-devices/ - FDA Guidance on AI-Enabled Devices: Transparency, Bias, Lifecycle Oversight. CenterWatch. October 29, 2025.
https://www.centerwatch.com/insights/fda-guidance-on-ai-enabled-devices-transparency-bias-lifecycle-oversight/ - Predetermined Change Control Plans: Guiding Principles. PMC. October 30, 2025.
https://pmc.ncbi.nlm.nih.gov/articles/PMC12577744/ - Step-by-Step ISO 42001 Implementation Guide. Axipro. May 15, 2025.
https://axipro.co/step-by-step-iso-42001-implementation-guide-axipro/ - SaMD Compliance Guide: Navigating Regulations for Software as a Medical Device. MDX CRO. October 7, 2025.
https://mdxcro.com/samd-compliance-guide-mdr-ai-act/ - Implications of the EU AI Act on medtech companies. JD Supra. July 16, 2024.
https://www.jdsupra.com/legalnews/implications-of-the-eu-ai-act-on-7851472/ - Bias Mitigation in Primary Health Care Artificial Intelligence Models. JMIR. January 6, 2025.
https://medinform.jmir.org/2025/1/e63642 - Bias in AI-based models for medical applications. PMC. June 13, 2023.
https://pmc.ncbi.nlm.nih.gov/articles/PMC10266078/ - Post-Market Surveillance for AI/ML-Enabled Medical Devices. FDA. September 28, 2023.
https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-aiml-enabled-medical-devices - 6 Key Steps to ISO 42001 Certification Explained. Cloud Security Alliance. January 15, 2026.
https://cloudsecurityalliance.org/blog/2026/01/15/6-key-steps-to-iso-42001-certification-explained - MDCG published new guidance on the interplay between the MDR/IVDR and the AI Act. Hogan Lovells. June 19, 2025.
https://www.hoganlovells.com/en/blogs/regulatory-radar/mdcg-published-new-guidance-on-the-interplay-between-the-mdr-ivdr-and-the-ai-act - Harmonised standards to the AI Act – for medical devices, too. Journal.fi. May 4, 2025.
https://journal.fi/finjehew/article/view/156791 - Artificial Intelligence-Enabled Device - Software Functions: Lifecycle Management. FDA. January 6, 2025.
https://www.fda.gov/regulatory-information/search-fda-guidance-documents/artificial-intelligence-enabled-device-software-functions-lifecycle-management - Subtle Medical Among First to Achieve ISO/IEC 42001:2023 Certification. Subtle Medical. July 29, 2025.
https://subtlemedical.com/subtle-medical-among-first-to-achieve-iso-iec-420012023-certification/ - ISO 13485: Key Updates on the Medical Device Regulatory Landscape. URM Consulting. November 23, 2025.
https://www.urm-consulting.co.uk/iso-13485-key-updates-on-the-medical-device-regulatory-landscape/
About The Authors:
 Srividya Narayanan, MDS,MSc, is a regulatory affairs specialist supporting global submissions for innovative cardiovascular and neurovascular medical devices. With an MDS in dentistry and a master of science in regulatory affairs from Northeastern University, she bridges clinical insight with regulatory strategy across AI/ML‑enabled devices, wearables, and real world evidence-driven clinical trials. She is the co‑founder of Path2RAQA, serves as an ambassador for ISPE and on the RAPS Events Committee, and has spoken at more than 30 global regulatory conferences. She can be reached on LinkedIn.
Srividya Narayanan, MDS,MSc, is a regulatory affairs specialist supporting global submissions for innovative cardiovascular and neurovascular medical devices. With an MDS in dentistry and a master of science in regulatory affairs from Northeastern University, she bridges clinical insight with regulatory strategy across AI/ML‑enabled devices, wearables, and real world evidence-driven clinical trials. She is the co‑founder of Path2RAQA, serves as an ambassador for ISPE and on the RAPS Events Committee, and has spoken at more than 30 global regulatory conferences. She can be reached on LinkedIn.
 Apekshit Mhatre, B.Pharm, MSc, is a regulatory affairs and quality‑focused life sciences professional with experience working in FDA/ICH‑regulated environments, where he has supported CMC operations, post‑market surveillance, and global regulatory submissions. He previously served as a drug safety and regulatory affairs coordinator at Fortrea, processing over 1,000 global safety cases and coordinating cross‑functional documentation and health authority submissions. Mhatre is currently pursuing a master of science in regulatory affairs at Northeastern University and an active Startup Boston and RAPS Event Committee member. He can be reached on LinkedIn.
Apekshit Mhatre, B.Pharm, MSc, is a regulatory affairs and quality‑focused life sciences professional with experience working in FDA/ICH‑regulated environments, where he has supported CMC operations, post‑market surveillance, and global regulatory submissions. He previously served as a drug safety and regulatory affairs coordinator at Fortrea, processing over 1,000 global safety cases and coordinating cross‑functional documentation and health authority submissions. Mhatre is currently pursuing a master of science in regulatory affairs at Northeastern University and an active Startup Boston and RAPS Event Committee member. He can be reached on LinkedIn.