
Clinical Investigation Plans: The Role Of MDCG 2024-3 In The Context Of EU MDR Annex XV, ISO 14155
By Leslie Hammermüller, Muna Kebede, and Matthias Fink

The Regulation (EU) 2017/745 (EU MDR)1 requires a pre-market clinical investigation for implantable and Class III medical devices unless one of the exemptions in Articles 61(4), 61(5), and 61(6) applies. The more stringent approach to demonstrating equivalence, especially to a medical device manufactured by a competitor, will likely increase the number of pre-market clinical investigations for new implantable and Class III devices.
Manufacturers conducting clinical investigations in at least one European Union (EU) member state must consider the relevant articles in Chapter VI and Annex XV of the EU MDR. Besides the legal requirements of the EU MDR, there is also the international standard ISO 14155:20202 on Good Clinical Practice for clinical investigations with medical devices.
The Medical Device Coordination Group (MDCG) published the 2024-3 “Guidance on content of the Clinical Investigation Plan for clinical investigations of medical devices” on March 12, 20243 on the content of the clinical investigation plan (CIP) for clinical investigations of medical devices.
This article demonstrates how MDCG 2024-3 links the legal text of the EU MDR with the ISO standard while guiding manufacturers on how to ensure alignment with the technical documentation and, particularly, the planned clinical evaluation of the subject device. It also highlights which document provides which specific information on the various aspects of the CIP.
How Does MDCG 2024-3 Complement Existing Guidance And Regulation?
Recital 64 of the EU MDR references ISO 14155 as the well-established guidance and internationally accepted standard for clinical investigations, which also helps non-EU clinical investigations conducted in accordance with the standard to be accepted within the EU. While the EU MDR is the legally binding document taking precedence over the requirements in a standard or a guidance document, all sponsors and/or manufacturers conducting clinical investigations within the EU are also expected to apply ISO 14155.
Manufacturers seeking additional guidance should always refer to the several guidance documents published by the MDCG, which is composed of representatives of all member states and chaired by a representative of the European Commission. Before the new 2024-3 document, the MDCG had already published six other guidance documents on various aspects of clinical investigations (CIs) in accordance with Regulation (EU) 2017/745 (EU MDR). These addressed safety reporting,4,5 application/notification,6 clinical investigation identifications (CIV-IDs),7 substantial modifications of the CIs,8 and an Investigator’s Brochure for clinical investigations of medical devices.9
Additionally, in December 2023, the updated MDCG 2021-6, Rev. 1, provided 35 questions and answers10 related to a CI. This MDCG document already referred to the, then still in a draft stage, MDCG 2024-3 in Question 29 on the expected content of a CIP.
These MDCG documents are not legally binding. However, since Notified Bodies (NB) are expected to follow MDCG documents, manufacturers should consider them to facilitate the conformity assessment process. Table 1 presents an overview of these three documents.
It should be noted that all three documents apply to medical devices and combination products, which contain a medicinal substance as an integral part. However, combination products must also follow further legal requirements in relation to the integrated medicinal product. Likewise, additional regulatory aspects might be relevant for multicenter clinical investigations partially conducted outside of the EU. MDCG 2024-3 provides guidance in addressing the extra burden.
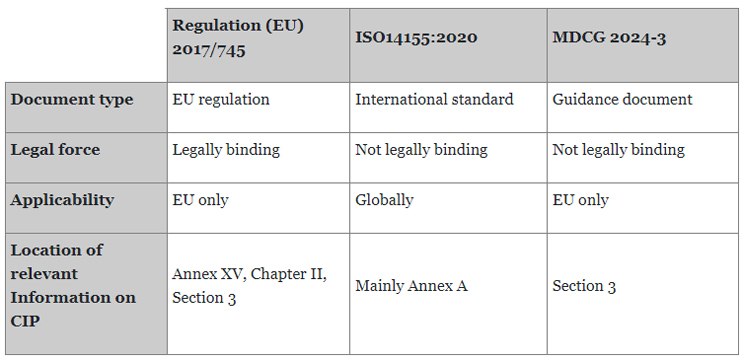
Table 1: Overview of the Regulation (EU) 2017/745, ISO14155:2020, and MDCG 2024-3
A CIP ensures that any planned clinical investigation complies with the current ethical and scientifically accepted state of the art on conducting a clinical investigation, ensuring good clinical practice. While the EU MDR and ISO 14155:2020 cover all relevant aspects of a clinical investigation, MDCG 2024-3 provides additional information and clarification on content of the CIP without introducing any new requirements.
Throughout the documentation, MDCG 2024-3 provides guidance on the content requirements as outlined in ISO 14155:2020 and aligns them with the EU-specific requirements for compliance with the EU MDR.
- Section 3 of MDCG 2024-3 specifically addresses the requirements outlined in the EU MDR Annex XV, Chapter II and covers each part of the CIP. It provides insight into the expected level of detail. There are 19 items listed as the minimum required content.
- Section 4 addresses additional EU-specific information that should be included in the CIP.
Key topics highlighted by the MDCG 2024-3 are presented below and summarized in Figure 1.

Figure 1: Schematic overview of the key aspects from MDCG 2024-3
Role Of The CIP Synopsis
Appendix A of MDCG 2024-3 provides the first official template for the synopsis of the CIP, which is a mandatory part of every CIP. This template enables a harmonized approach, facilitates the review, and eases the transfer of the content to the technical documentation, e.g., the clinical development plan. For ease of use, this template is not only attached as Annex to the main document but also available as separate word document.11
As noted in section 3.1, chapter II of Annex XV to the EU MDR, there are specific language requirements for the synopsis, which must be in an official EU language, and further requirements of concerned member states might apply.

Alignment With The Device Life Cycle And Technical Documentation
While ISO 14155:2020 focuses on the content and conduct of a clinical investigation, MDCG 2024-3 emphasizes the alignment of the CIP in relation to the device life cycle and the technical documentation. In particular, the following are specific to the guidance and are highlighted throughout:
- Premarket clinical investigations must consider the general safety and performance requirements (GSPRs) that require the support of clinical data — at a minimum, this would be GSPRs 1, 6, and 8.
- The device identification and description must align with the clinical evaluation process requirements.
- Any differences between the intended purpose, indications, and target patient population in the CIP compared to the future labelling of the device intended to be placed on the EU market must be carefully considered and justified to avoid being challenged by the NB.
- It emphasizes the need for consistency between the primary and secondary endpoints defined in CIP, the direct or indirect clinical benefits claimed by the manufacturer, and the safety and performance outcome parameters required for the clinical evaluation. This applies to both pre-market and post-market (post-CE-marking) clinical investigations.
Regional Differences
MDCG 2024-3 points out that the European Union is not a homogenous region, and regional differences may apply, e.g., regarding alternative treatment for the standard of care or differences in the healthcare systems in the various member states. Such regional differences must be adequately addressed in the CIP.
Risk Assessment
ISO14155:2020 follows a risk-based approach for planning and conducting a clinical investigation, which is mainly reflected in the procedural aspects, like the monitoring process. MDR 2024-3 points out that the EU MDR requires an even more exhaustive and comprehensive approach, including a risk-benefit analysis of the use of the device within the clinical investigation. The benefit-risk analysis needs to reflect the role of the clinical investigation within the whole clinical evaluation process. This includes considering post-market surveillance data where available and any similar devices, if applicable.
Formal Requirements
Clinical investigations conducted in the EU need to fulfil certain formal EU-specific requirements that are not applicable outside the EU:
- In addition to or in lieu of the CIP number often assigned by manufacturers, especially for global clinical investigations, manufacturers must request a CIP reference number, the CIV-ID.
- Specific manufacturer information that needs to be disclosed is listed.
- Reporting requirements must consider additional national requirements of the member state(s) in which the clinical investigation is conducted.
Key Takeaways
The key takeaways from MDCG 2024-3 and the answer to why this guidance document is relevant and why sponsors and manufacturers should consider it in addition to the EU MDR and ISO 14155:2020 are:
- The document helps manufacturers address the requirements set out by the regulatory framework (the EU MDR) and align with the requirements of good clinical practice when conducting a clinical investigation.
- Following MDCG 2024-3 supports compliance with the relevant requirements of the EU MDR regarding the CIP.
- MDCG 2024-3 bridges the gap between the EU MDR requirements and the current international ISO standard for EU-specific requirements.
- It provides an official template for the clinical investigation synopsis, which will facilitate the review by the competent authorities and the NB and could reduce the risk of questions during the conformity assessment procedure by the latter.
In summary, MDCG 2024-3 is a guidance document for manufacturers to meet EU MDR requirements while complying with ISO 14155 when creating a CIP for their device. It specifically targets clinical investigations conducted in the EU and emphasizes alignment with the technical documentation, particularly the clinical evaluation. It is also the first official document to provide a template for the CIP synopsis as an option, harmonizing the content and presentation of basic CI information throughout the EU.
References
- COMMISSION, T.E., REGULATION (EU) 2017/745 (EU MDR). 2017.
- Standardization, I.-I.O.f., Clinical investigation of medical devices for human subjects — Good clinical practice. 2020.
- MDCG, MDCG 2024-3 Guidance on content of the Clinical Investigation Plan for clinical investigations of medical devices. 2024.
- MDCG, MDCG 2020-10/1 Rev 1 Safety reporting in clinical investigations of medical devices under the Regulation (EU) 2017/745. 2022.
- MDCG, MDCG 2020-10/2 Rev 1 Guidance on safety reporting in clinical investigations Appendix: Clinical investigation summary safety report form. 2022.
- MDCG, MDCG 2021-8 Clinical investigation application/notification documents. 2021.
- MDCG, MDCG 2021-20 Instructions for generating CIV-ID for MDR Clinical Investigations. 2021.
- MDCG, MDCG 2021-28 Substantial modification of clinical investigation under Medical Device Regulation. 2021.
- MDCG, MDCG 2024-5 - guidance on content of the Investigator’s Brochure for clinical investigations of medical devices. 2024.
- MDCG, MDCG 2021-6 Rev. 1 Regulation (EU) 2017/745 – Questions & Answers regarding clinical investigation. 2023.
- MDCG, MDCG 2024-3 Appendix A Clinical Investigation Plan Synopsis Template. 2024.
 About The Authors:
About The Authors:
Leslie Hammermueller is a senior consultant and project manager at AKRA Team GmbH. After graduating in human biology, she was a clinical data manager for 10 years working on clinical studies in a global CRO before joining a medical device manufacturer as a clinical affairs manager. At AKRA Team, she supports manufacturers of all device classes fulfilling the requriements of the EU — on the operational level as well as providing in-depth training — with a strong focus on all clinical aspects.
 Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
 Matthias Fink, M.D., is a senior clinical consultant with AKRA Team GmbH. He provides consulting for medical device manufacturers of all sizes on regulatory and clinical requirements with a focus on the EU MDR. Previously, he worked as a clinical reviewer and manager of the Clinical Focus Team North America for TÜV SÜD Product Service in Germany and the U.S. A Board-certified orthopedic and trauma surgeon with 17 years of experience in orthopedic, trauma, and reconstructive surgery, as well as cardiovascular and thoracic surgery training, Fink is also a presenter at conferences and workshops on the clinical requirements and the implementation of the EU MDR.
Matthias Fink, M.D., is a senior clinical consultant with AKRA Team GmbH. He provides consulting for medical device manufacturers of all sizes on regulatory and clinical requirements with a focus on the EU MDR. Previously, he worked as a clinical reviewer and manager of the Clinical Focus Team North America for TÜV SÜD Product Service in Germany and the U.S. A Board-certified orthopedic and trauma surgeon with 17 years of experience in orthopedic, trauma, and reconstructive surgery, as well as cardiovascular and thoracic surgery training, Fink is also a presenter at conferences and workshops on the clinical requirements and the implementation of the EU MDR.