
Control Strategies For Injectable Drug Delivery Combination Products
By Steven Poon, principal consultant, 7TP Consultancy

When it comes to the development of pharmaceutical and biologic products, control strategies are well established. However, for drug delivery combination products, while the concept itself is not new, the definition and an overall plan for control strategies are just starting to catch on. ICH Q11 outlines the need to assure process performance and product quality (ICH Q10), but what does it mean for combination products and how can it be applied?
The two challenges and differences between drug and device development are: the definitions and the development pathway.
Critical quality attributes (CQAs) are clear for drugs/biologics but are less commonly used in device development. For drug delivery devices, in particular needle-based injection systems (NIS), primary functions1 (PF) and essential drug-delivery outputs2 (EDDO, or formerly essential performance requirements) are more commonly used terms, and they usually have an impact on:
- efficacy (in this case delivered dose),
- safety, and/or
- usability or intended use.
To strengthen alignment between drug and device teams, and to have more of an end-to-end view of controls, organizations should define “combination product CQAs” (red ellipse in Figure 1).
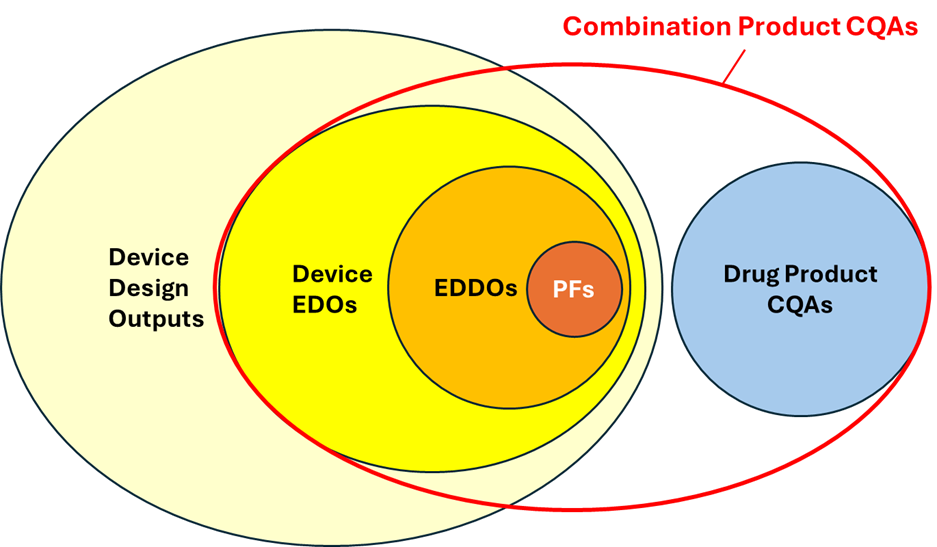
Figure 1: Combination product CQAs onion chart
PFs are usually also identified as EDDOs and are a subset of the essential design outputs (EDOs) and a totality of the development effort. In some cases, there’s an overlap between device CQAs and drug CQAs. Some examples include sterility, sub-visible particulates, or delivered dose, which can be applicable to both the drug product (in its container) and the drug delivery device. Understanding where overlap exists highlights the importance of an end-to-end control strategy starting at the beginning of development.
How To Identify Combination Product CQAs
The first step is to identify the system-level functions or operations, with focus on the combination product in the users’ hands as opposed to subsystem functions. Inputs into a system level function list include:
- the task analysis,
- primary operating functions,
- design input requirements,
- the intended use statement.
With this list of system functions defined, it is necessary to next assess and identify the PFs, EDDOs, and any other functions of the drug delivery device that could be deemed a CQA. The other functions can be identified by asking yourself this question: if they were to fail or degrade beyond the expected functional performance would that have an adverse effect on efficacy, safety or intended use?
Together, this is the list of device CQAs, which, along with the drug product CQAs (defined by drug product development/analytical) is your list of combination product CQAs. This forms the starting point of the control strategy: assuring product quality.
Control Strategy Planning: Known-Knowns And Known-Unknowns
One must accept that when establishing a control strategy plan, design output specifications, process controls, critical dimensions, process performance, or device performance may not be known; development is development. Therefore, the initial aim of a control strategy plan must be to identify:
- What needs to be controlled: the combination product CQAs (as defined above),
- What are the known-knowns: existing design or process controls (or lack thereof),
- What are the known-unknowns: additional design or process controls to be implemented, and verification evidence or validation data to be planned for.
This will inform potential design improvements, upstream and downstream process controls, parameters, or studies to be further characterized and understood, as well as informing design verification, validation, and process qualification plans. Much like risk management, the control strategy is a living process that is to be updated throughout development and monitored throughout the life cycle of the combination product.

Implementation of controls, upstream and downstream, can be best understood when illustrated by a funnel diagram (Figure 2). The further upstream the controls can be implemented, the more effective they generally are. Also, controlling subsystem/lower level CQAs on the device constituent and/or primary packaging component is generally more cost effective when compared to performing controls once the drug product has been added. In many cases, performance of the device constituent components or primary containers is not influenced by downstream processes, such as the final assembly process or presence of the drug product/container.
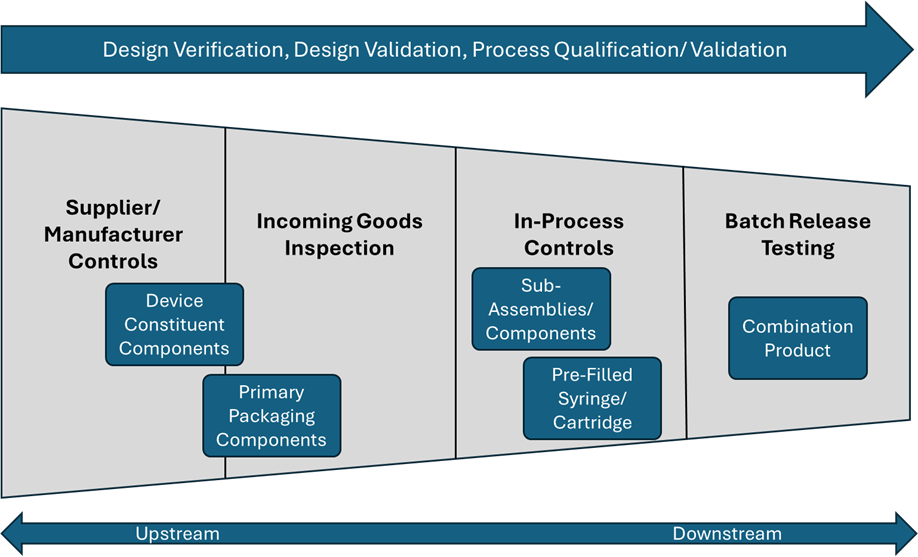
Figure 2: Funnel diagram of upstream vs. downstream controls
The types of controls, from upstream to downstream, can be categorized as follows:
- Supplier or manufacturer controls of the device constituent and/or primary packaging components. This includes control of critical or control dimensions, incoming controls of bought-in components, in-process manufacturing controls, and batch release testing.
- Incoming goods inspection of device constituent and/or primary packaging components. This includes review of appropriate certificates of analyses (CoA) or compliance (CoC).
- In-process controls during filling of the drug product into container (if applicable), the final assembly of the drug-device combination product, and packaging and labelling steps. This includes 100% in-line controls, in-process controls performed on samples of the batch during production, or controls that are part of the process design.
- Batch release testing of the combination product. This is typically a destructive test and reserved for combination product CQAs that are not adequately controlled in the other upstream processes or where proper controls cannot be applied upstream.
Alongside this, organizations should include and generate evidence through design verification testing, process qualification, and testing of product from the validation batches. In some cases, where a combination product CQA can be shown to be controlled as part of the design or material and is not influenced by the final assembly steps or presence of the drug product, further controls may not be necessary beyond ensuring that the combination product design and processes are unchanged. Where changes are experienced, standard change control procedures and re-verification or re-validation must demonstrate that the combination product CQA is still maintained.
To Release Or Not To Release: Importance And Integration Of Risk Management
As with much of device and combination product development, a risk-based approach must be applied. Consistent risk management should be implemented from the beginning of development, as it not only helps to identify the combination product CQAs, but also drives the organization’s strategy for design verification (sample size, acceptance criteria, statistical tolerance intervals), process qualification, and controls (incoming goods per ISO 2859-1, for example, release testing, etc.).
One’s appetite for risk is informed by data or evidence (or lack thereof), maturity, the complexity of the device constituent components and/or processes, and the potential harm to users, should something go wrong. As more data becomes available – through design verification testing, process qualification, etc. – the level of risk generally reduces (Figure 3). This data should be fed back into the control strategy in order to make informed decisions on the combination product CQAs, which will require testing on release of the finished product.
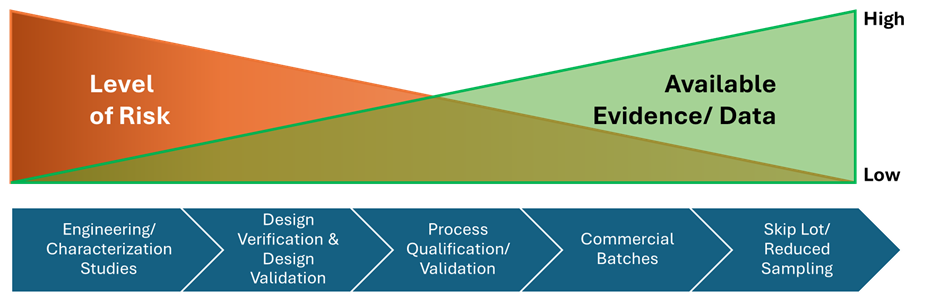
Figure 3: Risk vs. evidence profile through development
Just because a function or operation is identified as critical, a PF or EDDO, does not mean it must automatically be tested on release of batches. If it can be justified that a combination product CQA is satisfactorily controlled upstream in the design and/or process (and that there is evidence of such) or that it is not impacted by the downstream processes or components, then in most cases there is no need for control of this attribute at release.
Furthermore, as more data becomes available through the satisfactory lots produced, organizations should reassess (using a risk-based approach) adequacy of controls and sample sizes for release. Evaluation could potentially move toward skipped lots or reduced sampling, if the risk is low and acceptable.
In part 1 of this article, we outlined the importance of defining drug-device combination product CQAs, implementing a fluid control strategy, and understanding risk management throughout production and testing. In the second part of this article, we will provide an overview of the control strategy process paired with a case study.
References:
- As defined in ISO 11608-1:2022
- As defined in FDA’s draft guidance Essential Drug Delivery Outputs for Devices Intended to Deliver Drugs and Biological Products, June 2024, docket number FDA-2024-D-2560
About The Author:
 Steven Poon has been working in the medical device and drug-delivery space for over 15 years, with former roles in product development consultancy, OEM medical device manufacturer, and biopharmaceuticals. He often acts as a bridge between medical device development and drug product teams. Once described by a client as a “Swiss army knife”, Steven combines a background in mechanical engineering, a keen eye for quality, and a natural interest in technical and regulatory developments.
Steven Poon has been working in the medical device and drug-delivery space for over 15 years, with former roles in product development consultancy, OEM medical device manufacturer, and biopharmaceuticals. He often acts as a bridge between medical device development and drug product teams. Once described by a client as a “Swiss army knife”, Steven combines a background in mechanical engineering, a keen eye for quality, and a natural interest in technical and regulatory developments.