
3D Printing Production Medical Devices — Pitfalls And Best Practices
By David Schoon, Smithwise
Medical device developers use 3D printers religiously, to develop prototypes and to iterate designs, in order to rapidly learn and improve upon a product idea. Traditionally, these tools are used during the early stages of development. After prototyping is complete and the behaviors of a product or part design are understood and tested, the design is manufactured by a more economically viable method, such as injection molding, extrusion, casting, or metal stamping, amongst others.
However, there are circumstances under which 3D printing is a perfectly reasonable production method. These can be instances where production volumes are low, product margins are high, or there is a need to uniquely customize each design. While these may not be common scenarios for, say, a smartphone or a coffee mug, these factors can translate well to certain types of medical devices. Still, device makers need to be aware that there are precautions and processes to consider as a result of the unique risks associated with 3D printing manufacturing.
It’s likely that many developers with 3D printing exposure or experience already are aware of some of the common traits and risk associated with the process — FDM hole sizes that are significantly undersized, in accordance with claimed machine tolerances; photopolymerization materials that creep and dimensionally change over time when exposed to loading conditions; and DMLS parts that are saggy, droopy, or out-of-specification from the outset, due to poor build orientation and support material layout. However, these examples of common difficulties merely scratch the surface of what needs to be understood, from a risk perspective, when implementing 3D printing into a medical device design.
One difficulty that developers have encountered to this point is the particular dichotomy that exists between 3D printing — which can be associated with fast and loose development — and the regulatory standards and rigor imposed by FDA upon devices developed within a quality system. The ease with which one is able to generate parts through 3D printing inherently introduces risk.
In May 2016, the FDA released a draft guidance titled Technical Considerations for Additive Manufactured Devices. Any manufacturer or organization considering 3D-printed components during the development of a medical device should refer to this document. The guidance goes into detail regarding risk and other considerations related to 3D printing, as well as how to employ 3D printing within device development. Some of the risks and considerations discussed include:
- Development drawings with critical dimensions can be overlooked, and/or parts can be printed without files being saved and documented properly within a Device Master Record.
- Material behavior can vary significantly from datasheet specifications due to environmental conditions, build orientations, print machine variables, and other unanticipated factors.
- Software workflow, material controls, and post-processing are important considerations to achieve repeatable and quality parts, and proper documentation and manufacturing flow charts should be generated to capture these.
- Compared to traditional manufacturing techniques, there are feature size limits, dimensional variation based on technique, environmental conditions factors, and many factors affected by build orientations.
- Material behavior, with respect to cleaning and sterilization, can differ from that of parts manufactured using traditional methods.
The FDA has presented workflow guidance detailing what needs to be controlled for a successful device submission when utilizing 3D printed components. Developers should plan to include proper design documentation, software workflow, material controls, post-processing controls, and testing considerations.
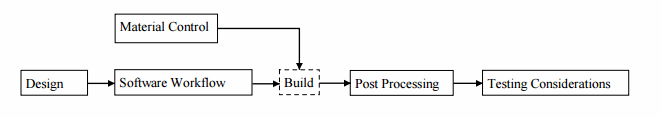
The guidance should be consulted for finer granularity regarding each of these quality components. Additionally, manufacturers should heed these commonly overlooked considerations relevant to 3D-printed part production:
- It is recommended that performance verification come from testing finished parts, or coupons that are produced using an identical process.
- If possible, device files should be maintained and archived to an Additive Manufacturing File format, as described in ISO/AST 52915.
- Workflows should be established to include part placement, layer thicknesses, printer accuracy, print speed, and the build layout within a print envelope.
- Printer maintenance procedures should exist along with workflows to establish consistency between builds. These should be maintained within the Device Master Records (DMR).
- All material information should be documented, including any process aids, like material support and crosslinkers.
- Workflows should exist for any post-processing steps, such as the removal of support material. Depending on the use case, testing may be necessary to understand the effect this action may have on the finished part.
- Process validation should be established. This can include monitoring and documenting the 3D printer’s environmental conditions to validate the machine process.
The FDA guidance is intended to induce a thoughtful approach that will yield a successful regulatory submission, and is catered towards the inclusion of 3D printed components within an end product. For a low-volume FDA evaluation or a clinical study, there may be economic factors that make 3D printing a desirable approach for component and prototyping purposes. When weighing this approach, it should be understood that the burden is on the developer to establish sufficient rationale to claim equivalency between a prototype and a high-volume production method.
In certain instances, equivalencies can be rationalized fairly simply — for example, if a 3D-printed housing was used for a laparoscopic disposable within a human factors validation study. However, if said device contained electrical or antenna components and was being used within a clinical trial or certification testing, there may be good reason to prototype with a production material. Material differences could influence the EMC or antenna performance, and having to retest to IEC 60601 conformity could be costly and time-consuming.
In summary, many factors need to be weighed when implementing 3D printing within an evaluation, clinical, or production device setting. Because 3D printing is as easy as a click of a mouse, additional responsibility falls on developers to properly design, assess risk, or account for quality. As the FDA has only recently began to weigh in on 3D printing, developers and manufacturers would be wise to use pre-submissions when clarifications are needed.
About The Author
Dave Schoon is the Director of Mechanical Engineering at Smithwise's Newton office. Dave leads the technical team while maintaining an active hand in ongoing projects. His recent projects include a machine for sleep apnea sufferers, a pill-dispensing robot, and a handheld audiology device. Prior to joining Smithwise, Dave was a mechanical engineer with Datum3D where his notable projects included development of an airborne pathogen detector and an endoscopic performance validation machine. He also developed a number of medical devices and disposables with a focus on DFMA and cost reduction, including minimally invasive biopsy instrumentation and endoscopes. In addition to Dave’s professional experience, he served as a mentor for product development courses at MIT and hosted Top Down Design workshops in SolidWorks. Dave holds a B.S. in Mechanical Engineering from Worcester Polytechnic Institute.