
Documentation Priority For Early Stage Medtechs — Part 1: User Specification
By Tommy Cooper, Cooper Consulting Service

A common dilemma for early stage medical device companies is their lack of expertise in medical device development. As expected, the development team is busy establishing the company itself while still trying to define the product and answer questions like:
- What are the product’s essential features?
- Does the technology work consistently?
- What makes our product better than the competition?
- Does the draft business plan make sense?
- What is my funding strategy?
- What is my market?
- How will my product be used?
While trying to respond to these questions, the company is being advised of the significant effect documentation, regulatory strategy, and quality management have on company value. This article addresses documentation the company should prepare while dealing with other project startup questions and issues.
Many startups focus on designing proof-of-concept prototypes and operate without a formal quality management system (QMS). They do not have standard operating procedures (SOPs) or formal documentation systems. Even so, they should prioritize the following planning and high-level requirements documents:
- User Requirement Specification (URS), or Product Requirement Specification (PRS)
- List of Applicable Documents (Standards)
- Block Diagrams
- Project Development Plan (PDP)
- Project organization and management responsibility
- QA Plan
- Regulatory Plan
- Engineering development plan and schedule
- Software Development Plan (SDP)
- Usability Plan
- Feasibility Test Reports
- Initial Hazardous Situation List (HSL)
These documents define a project’s scope and improve communication between the development team, outside vendors, management, potential users, and investors. They also organize the project and address some issues that may arise as the project begins. Finally, these documents enhance the development team's effectiveness and should not be considered a distraction.
User Requirement Specification (URS) / Product Requirement Specification (PRS)
Depending on the type of product and individual preference, these requirements may be separated into two documents: one focused on the user, and the other centered on the device. Or, they can be integrated into a combined URS/PRS document. This segment of the article describes sections of the URS/PRS.
Scope
The scope defines which devices are covered by the specification document. For example, an extracorporeal blood filtering system may consist of three separate assemblies: the flow control unit, the disposable tubing kit, and the blood filtration cartridge. A wireless ECG/respiratory monitoring system may consist of a patient module, a disposable electrode set, and a software application (app) on a mobile device to display the data.
Reference Documents
Reference documents should include the list of applicable standards, along with any other feasibility reports and the user requirements overview. The list of applicable standards — to be covered in a later article in this series — should be maintained as a separate document. For now, here is a quick overview of some sources for the list of applicable standards:
- FDA Recognized Consensus Standards
- FDA Guidance Documents
- EU Harmonized Standards List for Medical Devices
- EU MDR Common Specifications (ex: Reprocessing of single-use medical devices)
- EU MDR Guidance Documents
- EU Directives
- Health Canada List of Recognized Standards for Medical Devices
- Health Canada Guidance Documents
A list of any abbreviations should also be included in the reference documents section.
Intended Use
Intended use is specifically referenced in FDA regulatory applications and determines how the device will be classified and treated by regulatory agencies. A good way to draft the intended use statement is to use the FDA 510(k)/De Novo/PMA summary from a similar product. The associated databases for the summaries — the 510k Database, the De Novo Database, and the PMA Database — are available online.
A good initial approach is to select predicate devices similar to the product and to incorporate their intended use into your requirements document. In submitting a 510(k), the goal is to match the predicate device’s intended use as closely as possible. For the De Novo route, it will be necessary to prove why your device is not substantially equivalent to other similar devices, and to indicate why your device is not Class III. A PMA (Class III) submission will require knowledge of any alternative practices or procedures for diagnosing, treating, preventing, curing, or mitigating the disease or condition for which the device is intended, since there is not a substantially equivalent device.
Before talking with investors or making external presentations, it may be helpful to discuss the product with a few regulatory professionals to make sure your early regulatory strategies are plausible. If the product is designated for non-510(k) pathways, the regulatory process will be more expensive and will require additional submission material. However, this could offer a unique market advantage, in that 510(k) devices already have "substantially equivalent" competitors, while De Novo and PMA devices do not.
Note: When you are identifying similar devices, make sure to save a copy of their submission summary, their FDA classification product code, and their regulation number.
Classification
The classification of the medical device per IEC/EN/ES 60601-1 — the basic safety and essential performance standard — is identified in this section. The primary categories are:
- Class I or Class II electric shock protection — Class I is powered from alternating current (AC) mains and has a connection to protective earth (ground) as part of the protection. Class II may be powered from AC mains or internally powered, but does not have a connection to protective earth and has at least two layers of protective insulation (double insulated).
- Type B, BF, CF, and defibrillation-proof applied part protection — Type B is for patient-applied parts that are not electrically isolated. Type BF is for patient-applied parts that are electrically isolated and are rated for contact with the skin. Type CF is for electrically isolated, patient-applied parts that are rated for connection to the heart or blood vessels. Defibrillation proofing may be required for type B, BF or CF applied parts.
- Handheld, body-worn, portable, mobile, or stationary operation
- Single patient use or reusable
- Sterile or non-sterile
These classifications relate to how the equipment is used and to safety testing requirements. Some medical device standards may require specific classifications; for instance, a Type CF applied part with defibrillation protection is designated in the ECG standards. Knowing these classifications early in the development process assists with device design decisions.
Environmental Conditions
Devices need specifications for operating conditions and storage/transport conditions, based on the intended use environment. Below is a standard set of operating, storage, and transport conditions that would cover a range of use environments.
- Operating Environment:
- Temperature: +10°C to +40°C
- Humidity: 30% to 75% (non-condensing)
- Pressure: 70 kPa to 106 kPa
- Altitude: less than 2,000m
- Storage/Transport:
- Temperature: -40°C to +70°C
- Humidity: 10% to 98%
- Pressure: 50 kPa to 106 kPa
- Altitude: Suitable for air transport
The operating environment is based on factors including the ratings for selected components, the specifications for competitive devices and, most importantly, the actual environment for your device. For example, if you expect to sell the unit worldwide, some countries may have significant populations in higher altitudes, requiring more stringent dielectric withstand specifications.
Remember, these are system operating requirements and the temperature inside your device enclosure may be much higher than the ambient temperature due to heat dissipation in internal components. A higher upper operating temperature may be necessary for EMS use outdoors, or for use in hospitals without air conditioning. Batteries are particularly sensitive to high temperatures — which can reduce battery life and cause battery swelling/leaks — and to low temperatures, which can reduce battery capacity.
Additionally, specialized storage and transport requirements may lead to special handling procedures for carriers; a wide temperature range covers conditions that might be encountered when a device is in a shipping truck during a hot summer day or a cold winter day. If the device cannot withstand a range of temperature or humidity conditions during shipping, it may require special packaging and physical indicators, or special handling instructions, resulting in higher shipping costs.
Likewise, some devices may be affected by shock or vibration and need special protective packaging to ensure that shock limits have not been exceeded. Batteries may require special handling, too, as certain batteries cannot be shipped by air (due to the changes in pressure) or may be damaged by high temperature. For example, a biological absorptive material may be less efficacious if exposed to conditions outside a specific, narrow range.
Use Environment
Defining use environment means answering the following questions:
- What are the devices’ use scenarios? Think through the steps for device use (on a patient) in normally foreseeable situations and locations.
- Is the device handheld, portable, body-worn, mobile, or stationary?
- In what specific areas /conditions should the device be stored, transported, staged (if necessary), and used?
- Will the device be subjected to fluids, such as IV solution, bodily fluids, cleaning agents, sweat, showering, etc.?
- Will the device be used in areas without dust control, such as outdoors or in a patient’s home?
- Will the device be used near an MRI, an electrosurgical unit, a cell phone, or another source of electromagnetic interference?
- Will the device be used in an oxygen-rich environment, or near flammable anesthetics or other flammable gases?
- How long will the device be in use for a treatment session? How many treatment sessions per week?
- Does the device require reprocessing or disinfection between uses?
- For sterilized single-patient-use devices, what sterilization processes will be used?
- For battery powered devices, how long is a treatment session? How many treatments are done per day? How long should the battery last before replacement?
- For rechargeable devices, what is the battery operating time? How long does it take to recharge the device between uses? Can it be charged during use? Is battery operation a normal condition or strictly emergency back-up? Does the battery need a minimum charge level to allow the device to be used?
- What are the facility requirements for installation of the device?
- How often will the device be used (e.g., daily, once a month, or with irregular frequency)?
- Will the user will receive training? If so, how often will it need refreshed? Who conducts the training (device company personnel, qualified staff, etc.)?
- Will the user operate other devices or attend to other patients while using this device?
- Will the device’s therapy period extend past a nursing shift change?
- Will a patient with a home-use device sleep with the device in operation?
Users
Different user group profiles for the medical device must be defined to indicate users’ mental, physical, and demographic traits, as well as any special characteristics, such as occupational skills, job requirements, and working conditions. User groups may be separated as patients, physicians, nurses, medical technicians, service technicians, biomedical technicians, home caregivers, and others.
The attributes by which these user groups are separated — such as age, culture, expertise, or type of interaction with the device — may influence device design requirements. Key user group parameters that must be defined include:
- Education/Certification — Educational requirements may include a college degree or special certification [e.g., MD, RN, Certified Biomedical Equipment Technician (CBET), Certified Clinical Perfusionist (CCP), Certified Clinical Hemodialysis Technician (CCHT), Certified Respiratory Therapist (CRT), etc.]
- Training — Whether provided by a college, the hospital, or the device manufacturer, training may be a requirement of some user groups. In some cases, the training may require learners to earn a certificate or return regularly for continuing education.
- Experience — For some equipment, users may need several years of experience as a resident or intern before securing clearance to operate the device unsupervised.
- Conditions — The allowable conditions for each group need to be defined:
- Trained within the last 12 months
- Trained but only operate the device once a month
- Certified company trained service technician
- Certified company trained operator
- Require verified identification or password
- Demographics — Some groups may have limitations on age, physical size, strength, visual acuity, color blindness, hearing acuity, etc. Examples include:
- A home-use monitoring product, wherein the patient is the user and may interact with a home care nurse to apply the device or be trained in its use. A secondary user may be the nurse or doctor who monitors the device output via a remote connection.
- The user for an X-ray device may be a radiological technician, nurse, or physician who directly modifies imaging parameters and positions the device to acquire desired views of specific anatomical areas. A primary user will be the radiologist who views and interprets the output images. X-ray devices output dangerous levels of radiation and thus require user identification verification before operation.
- The clinical technician user receives extensive training to perform 3D ultrasound procedures for cardiac assessment, in which results are interpreted by a cardiologist user.
- For a fetal monitor used to monitor patients in active labor, the Ob/Gyn-trained nurse user applies sensors to the patient and acquires the fetal heart rate and uterine contraction waveforms, which are interpreted by the Ob/Gyn physician and nurse to manage the patient.
- The user may also be company trained field service technicians who install, maintain, and service the device.
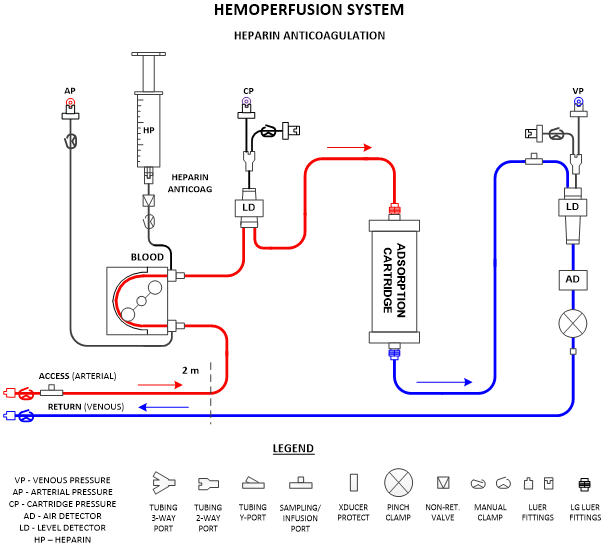
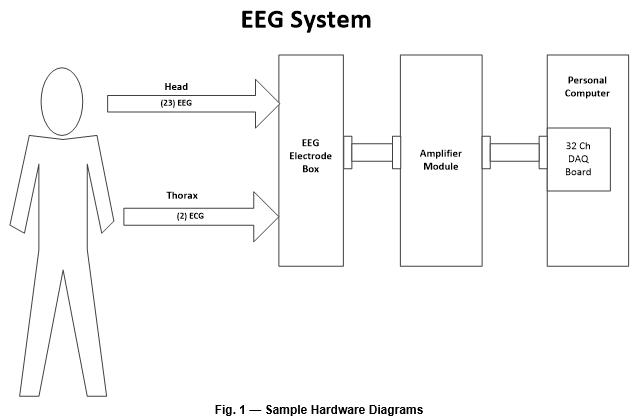
System Description
This section presents the device usage and major components at a high level and includes a block diagram of the system’s major components (Fig. 1). If the device features different configurations or different use modes, they can be diagramed and explained in this section. This section should make the device operation and components understandable to non-technical individuals, such as investors, executives, and regulatory personnel.
General Design Requirements
In this section, general requirements for the system’s design, operational phases, and usability requirements are described. General design requirements cover a range of system features. For example:
- System display must be a minimum 9" diagonal with touchscreen operation
- Display status must be readable from a specified distance of 10 m and viewable over a specified angle
- System housing must be designed with no difficult-to-clean crevices or edges
- Mobile systems must be stable on a specified incline during use and transport, must have brakes to prevent unexpected movement, and must have positioning handles to move the cart.
- Portable or handheld systems must be constructed to withstand the mechanical shock resultant of a drop from a specified height and still operate safely
- System must have a means to store cables during operation and transport
- System size must be limited to X/Y/Z dimensions due to limited space in patient area
- The alarm system requirements must be defined for alarm priority, audible, visual and tactile indicators, muting, clearing, and displaying the description of alarm conditions and actions for resolution.
- Device life cycle, including minimum number of years of expected operation and any required warranty
- Language management requirements depend on expected markets. The ability to localize languages has a significant impact on Graphical User Interface (GUI) design. The specification may designate a language for initial release with capability for localization. For example, the requirement may be for initial release in English (US) with GUI designed for localization of languages in future markets.
- Disinfection and/or sterilization requirements should be specified to help with selection of disposable accessory materials and exposed device materials.
- Product configurations or subassemblies, as well as combinations of accessories for the device
System Operational and Usability Requirements
These requirements usually focus on phases of system usage, such as power-on test, setup and initialization, therapy delivery, post-therapy, and storage. Here are example requirements:
- System must be designed for ease of servicing and access to internal components
- Time limit for setup, which may include power on, self-test, disposable kit installation, priming, and readiness verification.
- Safety testing requirements at startup and at periodic intervals
- Safety system visual and audible indicators
- Instructions for use must be available on the user interface
- System must have a maintenance-only login mode for service personnel
- Safety features that are required during therapy:
- Pressure measurement system to prevent blowout in fluid management system
- Arterial filter to prevent clots in blood pathway and trap bubbles
- Control switch IPXX rating for moisture and particle ingress to prevent unexpected operation
- Data Interface requirements for the device, such as DICOM transmission, USB port for system software updates, and Bluetooth for wireless data transfer
Note: There can be overlap between general design requirements and system operation requirements. It is not a black-and-white divide, but two different ways of thinking through high-level requirements.
Performance Requirements
Performance requirements define quantifiable specifications that are testable. A particular sub-component or sub-system that will not have separate component requirements may be specified in this section. Here are some examples:
- Total enclosure size
- Device weight
- Flow rate range and accuracy
- Applied energy limits, such as a TENS device or electrosurgical unit
- Mechanical rotation, translation, bending ranges, and position accuracies
- Pressure channel measurement range and accuracy
- Power input limits
- Battery related requirements
- Primary cells or rechargeable
- Battery operating time
- Recharge time
- Is it safe for battery to be recharged during patient use?
- Is battery operation a normal condition or strictly emergency back-up?
- Does the battery need a minimum charge level to allow the device to be used?
- Power draw during standby and operation
- Speaker output requirements for alarms
- Temperature limits for internal components
User Interface Requirements
User interface (UI) requirements should specify all UIs on the device to guide the usability plan’s outline. UI requirements can include things like hard keys, indicators, graphical user interface, and transport/wear interfaces. Here are some examples:
- Hard Keys and their associated functions
- On/Off switch — power the device on/off with momentary push or instant response
- Hand switch/foot switch — activate energy output
- Tubing clamp open/close switch
- Emergency stop switch — disable all hazardous outputs or stop all flow control devices
- Keyboard — data entry
- Indicators and their associated functions
- Status lights — standard green (normal), yellow (warning), and red (alarm)
- Battery charge — charging status
- Communications connection status
- Energy output — indicate hazardous energy output in progress
- Audible — indicate on/off, energy output, alarm, etc.
- Graphical user interface
- Sensor output status
- IFUs can be displayed here
- Menu navigation limited to three menus in depth or less
- Data entry for patient info will allow for user confirmation of entry
- Reset function for alarms and warnings
- Online time-stamped event/setting history page
- Transport/wear/operation interfaces and their associated functions
- Brake release — allow device mobility
- Foot brake — stop device mobility
- Handles — mechanical interfaces to apply moving force
- Elastic band — hold sensor package in place over anatomy of interest
Labeling, Packaging, Transport, and Storage
Labeling requirements will vary depending on potential markets. Here is the basic information included on device labeling:
- Manufacturer address, phone number, and email
- Product name
- Model number and revision
- Serial number and/or lot number
- Date of manufacture and/or expiration date
- Power requirements (voltage, frequency, watts, current)
- IP rating
- Fuse rating
- Applied part rating
- Sterilization
- Re-use
- IFU Symbol + eIFU http
- UDI (currently in the US and EU)
Depending on risk management mitigations, usability studies, EU MDR requirements, FDA guidance, and other (i.e., country specific) standards, additional labeling may be required. The product labeling material must withstand durability and environmental testing, and the text must be “clearly legible,” according to standard definition.
In addition to device labeling, device packaging requires labeling. This labeling contains similar information to the device labeling but includes transportation-specific information:
- Manufacturer address, phone number, and email
- Product Name
- Model number and revision
- Serial number and/or lot number
- Date of manufacture and/or expiration date
- UDI (currently in the US and EU)
- Transport temperature range
- Transport humidity range
- Transport pressure range
- Air transport restrictions
- Sterilization
- Re-use
- IFU Symbol + eIFU http
- Hazardous transport labeling
Depending on the transport environmental specification, rough handling limits, and device components, packaging may require features like humidity protection or sensors to detect whether a force limit has been exceeded. If the device is sterile, the packaging will have to pass standards testing and have a specified shelf life for sterilization. If a device requires limitations on transportation environment, it may require special handling for storage and transport, such as temperature-controlled storage, ground/sea transport only, and/or flammability precautions.
Product Documentation
These requirements usually are focused on manuals that will be needed (e.g., operator and service/install), whether the manual will be paper-based only or both paper-based and electronic (on the device), and languages into which the manual will be translated. Depending on the user’s education level and UI complexity, the user manual may require videos. Note that EU MDR requires electronic manuals.
Training/Services
These requirements indicate how the company will train users and service personnel. For example, service staff and application specialists will be required to attend training courses at the company, while medical staff may be trained by the application specialists. This documentation also identifies what training materials need to be developed.
Disposal
Depending on the device’s target market(s), disposal requirements may differ significantly. A common theme for specialty disposal is that local regulations should be followed for batteries. It also is common that materials used in the machine should be recycled if possible.
If the device will be marketed in the EU, disposal requirements will include compliance to the Restrictions on Hazardous Substances (RoHS) and the Waste Electrical and Electronic Equipment (WEEE) directives. ROHS also applies to devices that will eventually be marketed in China and other countries.
Conclusion
The URS/PRS is a key document in the product development process and should be prepared and released during the product’s initial development phase. For simple projects, this documentation may be prepared in the first few months. For complex products with several subsystems, it may take up to a year to draft and approve the URS/PRS.
The effort to prepare this document will clarify device features and key performance characteristics. Having this document, along with the others listed at the beginning of this article, will add value and organize the product development process, an important factor in the success of any startup.
About The Author
Tommy Cooper, PE has a BSEE ’74 and MSEE ’81 from University of Houston. He has worked in the medical device industry for over 40 years. He served 12 years as the co-chairman of the AAMI Blood Pressure Transducer Committee, holds 22 patents in the field of medical devices, and has published more than 16 articles in professional journals. He has developed numerous medical devices from concept phase to regulatory approval and production. Tommy founded Cooper Consulting Service (CCS) — an engineering design and development company with a focus on medical devices located in Friendswood, TX — in 1986.