
EU MDR Now Regulates Beauty Devices As Medical Devices
By Hilde Viroux, PA Consulting

The beauty devices business is booming. The global value of the cosmetic industry amounted to over $530 billion in 2022, with an expected growth rate of roughly 4% over the next five years, while the beauty devices market size stood at $66 billion and a growth rate over 20% through 2030.
Beauty devices are products that are used on the face and body for cosmetic reasons, such as, for example, hair removal devices, liposuction devices, and colored contact lenses. The access to the European markets has been easy for these devices as the Cosmetics Regulation 1223/2009 does not apply to devices, since this regulation only applies to pastes, creams, and lotion types of products.
The business model for beauty devices is often based on the “white label” model, in which distributors buy the product from a manufacturer and put their own name and branding on it.
What Are The EU MDR Requirements?
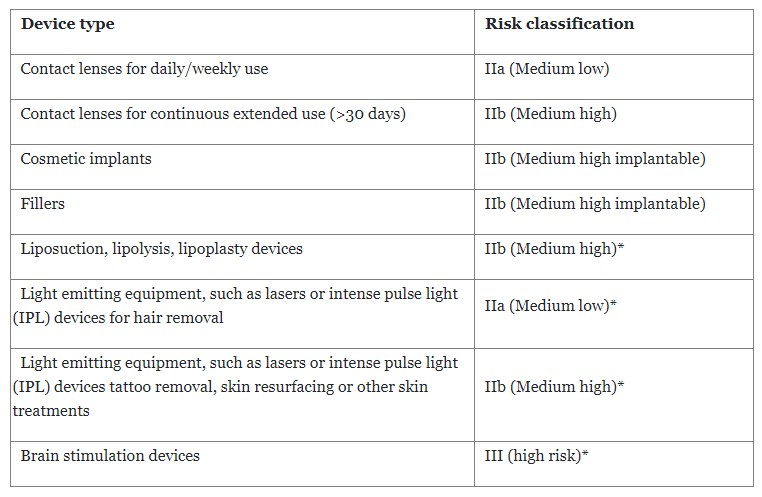
The EU Regulation 745/2017 on medical devices (EU MDR) lists in Annex XVI several beauty devices that the EU Commission wants to regulate as medical devices. A key element for a product to qualify as a medical device in the EU is that it must have a clinical benefit. This clinical benefit must outweigh the risks associated with the use of the medical device. Beauty devices don’t have a clinical benefit, as reducing wrinkles or having smoother skin is not considered a clinical benefit. The EU Commission has now defined ‘Common Specifications” in the implementing regulation 2022/2346, which gives requirements for the risk management and labeling for each of the following specified groups of beauty devices:
- Contact lenses, e.g., the so-called fun lenses, nonprescription colored contact lenses
- Cosmetic implants (apart from breast implants, which are already considered medical devices)
- Cosmetic fillers, e.g., products injected to plump up lips, reduce wrinkles
- Liposuction, lipolysis, and lipoplasty devices
- Light emitting equipment, such as lasers or intense pulse light (IPL) devices for hair or tattoo removal, skin resurfacing, or other skin treatments
- Brain stimulation devices and devices that apply electrical currents or electromagnetic fields that penetrate the cranium to modify neuronal activity, e.g., to stimulate or influence moods
The devices get a risk classification per Annex VIII of EU MDR. However, Regulation 2022/2347 defines the risk classification for some of the devices where the classification rules from the EU MDR may not be clear (marked below with *).
With the inclusion of these devices under EU MDR, manufacturers not only need to apply the elements of risk management and labeling as specified in the Common Specification but also need to comply with all elements of EU MDR — in other words, become de facto a medical devices manufacturers.
However, manufacturers have some time to become compliant, based on the risk classification of the device and depending on whether they have to conduct a clinical investigation. The latter will be the case for devices that are high risk (brain stimulation) or implantable (fillers, cosmetic implants).
What Are The Key Timelines?
The regulation provides a definition of the key timelines for compliance, depending on the type of product:
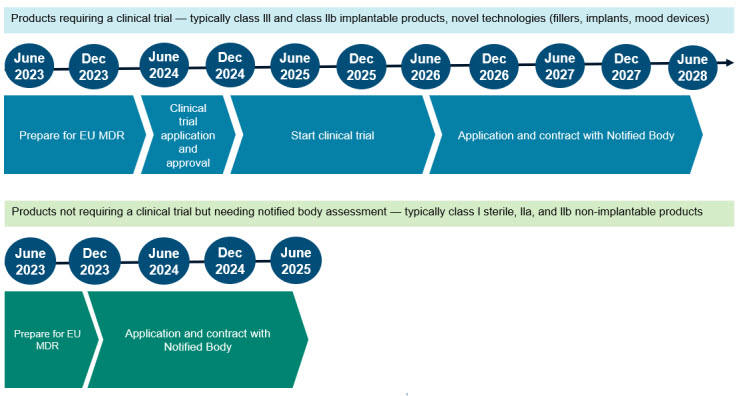
Devices that need a clinical trial as part of the approval procedure, have until June 2028 for compliance, with the caveat that (1) the clinical trial application must be approved before December 2024, (2) the clinical investigation is started before June 2026, and (3) a contract with a notified body is in place by June 2028. If any of the due dates are not met, the device may no longer be sold in the EU.
For devices that don’t need a clinical trial but do need the involvement of a notified body for CE marking, the timelines are significantly shorter. The manufacturer has between September 2023 and June 2025 to initiate the conformity assessment with a notified body.
What Are The Challenges For Manufacturers?
December 2024 may seem like plenty of time to obtain approval for a clinical trial application. However, before the application can be submitted, the design documentation and risk management file must be completed, and an investigator brochure and clinical trial application must be prepared and submitted. This will require new expertise, either in-house or external. Two years seems a short time for completing this, considering that the competent authority in-country also needs time to review and approve the application. A competent authority that has no experience with beauty devices may need more time than usual for review and approval.
But this is only a part of the compliance exercise. Manufacturers of these devices must fully comply with all requirements of EU MDR. This means a quality management system (QMS) that is in line with ISO 13485, a design history file for the product, fully compliant technical documentation, and proactive post market surveillance. This is not an easy undertaking, as evidenced by the large number of manufacturers of regular medical devices that are not compliant with EU MDR (per Team-NB Sector Survey report, May 2022).
Becoming compliant with EU MDR is more than just updating documents for regulatory compliance. It strengthens the device development processes by requiring a design history file for all products. It elevates clinical data requirements throughout the life cycle of the device. And it requires manufacturers to proactively monitor the safety and performance of the devices in the market. In addition, compliance brings the supply chain into the scope of the quality management system.
Manufacturers of beauty devices that also have medical devices in their product portfolio have the advantage of understanding the EU MDR requirements and may already have a relationship established with a notified body. Manufacturers that currently don’t have a contract with a notified body may experience difficulty contracting with one due to the limited bandwidth of EU MDR designated notified bodies and due to the fact that not all notified bodies will have these beauty devices in scope for certification.
Distributors that sell beauty devices under their own brand name may have to reconsider their business model, since they may become the legal manufacturer of the device under EU MDR and, as a result, they must meet all the requirements for a medical device manufacturer.
Many beauty devices are sold directly to consumers. Design changes and features enhancements tend to be frequent for those products to cater to the consumers’ interest and to stay ahead of the competition. However, the regulation 2022/2346 does not allow significant design changes to the product from June 2023 until the product is EU MDR compliant. This development constraint may have an impact on the manufacturer’s ability to introduce new product enhancements.
What Should Device Manufacturers Do To Get Ready For The EU MDR?
Manufacturers of beauty devices should start planning for EU MDR compliance. As a first step, begin with a thorough gap assessment of quality management system processes and product documentation, which will provide an indication of the remediation effort needed to support compliance. The assessment will support a review of the product portfolio; the objective of this activity will be to analyze the cost impact of compliance on revenue by product.
For the products that a manufacturer wants to maintain on the market, an implementation plan must be developed considering compliance timelines, key milestones, and organizational impact on available resources.
Distributors that sell beauty devices — impacted by the EU MDR regulation — under their own brand name must evaluate whether they want to take on the responsibilities of a medical devices manufacturer. Alternatively, distributors may choose to change their business model in order to meet the new EU MDR requirements.
About The Author:
 Hilde Viroux is a medtech expert at PA Consulting and is an expert on the European Medical Devices Regulation. She has a broad experience in regulations, quality, manufacturing, supply chain, and project management in the pharmaceutical and medical device industry. She has an MSc in medical technology regulatory affairs from Cranfield University in the U.K. and a BS in biochemistry engineering. Connect with her on LinkedIn.
Hilde Viroux is a medtech expert at PA Consulting and is an expert on the European Medical Devices Regulation. She has a broad experience in regulations, quality, manufacturing, supply chain, and project management in the pharmaceutical and medical device industry. She has an MSc in medical technology regulatory affairs from Cranfield University in the U.K. and a BS in biochemistry engineering. Connect with her on LinkedIn.