
EU MDR: When Products Without An Intended Medical Purpose Meet Medicinal Substances
By Muna Kebede and Matthias Fink, AKRA Team GmbH

One major change introduced by the Regulation (EU) 2017/745 (Medical Device Regulation [MDR]), compared to the former Directives 90/385/EEC (Active Implantable Medical Device Directive [AIMDD]) and 93/42/EEC (Medical Device Directive [MDD]), is the inclusion of products without an intended medical purpose, as listed in Annex XVI of the EU MDR (refer to Table 1 below). This addition was introduced to close a regulatory gap for products without an intended medical purpose but with similar risk profiles to medical devices. These include aesthetic and lifestyle products like colored contact lenses or dermal fillers, which have caused serious harm to users.1,2 Most major regulatory jurisdictions do not separately identify such products covered by Annex XVI of the EU MDR but include them under the existing regulations for medical devices. Other regions may regulate them as consumer products depending on the claims.

Table 1: Overview of the groups according to sections 1–6 of Annex XVI to EU MDR with product examples and the annexes of the Regulation (EU) 2022/2346 (CS) that set the specific requirements.
The terminology “without an intended medical purpose” does not necessarily reflect a lower risk or clinical relevance when compared to products with a medical purpose. The regulatory considerations for some of these aesthetic products, such as dermal fillers, also become more complex once they straddle multiple regulatory frameworks. Annex XVI products utilized in aesthetic procedures can contain medicinal substances (e.g., lidocaine hydrochloride [HCL]3), animal tissue (e.g., bovine collagen), or substances of animal origin (avian hyaluronic acid) or can include a treatment where the substance is derived from human blood (autologous). Table 2 below presents an overview of dermal filler product examples and additional applicable legislation that must be considered. Besides considering the additional legislation, manufacturers of products without an intended medical purpose must also apply a set of harmonized risk management and clinical evaluation requirements specific to their Annex XVI group (Table 1). These are defined in the Regulation (EU) 2022/2346 setting the common specifications (CS)4 that provide further product specific and legally binding criteria.
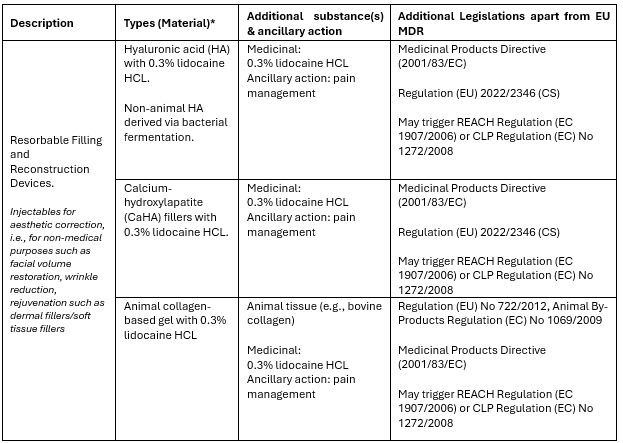
Table 2: Overview of dermal filler product examples (an Annex XVI product) as combination products.
*Material impacting the primary mode of action: physical/mechanical action, and not pharmacological, immunological, or metabolic.
In addition to the CS, manufacturers of such products must carefully navigate the diverse set of regulatory intersections under consideration of several guidance documents on:
- the differentiation between a medical device and other types of products and their classification5,6
- consultation procedures and their documentation,7,8,9 as well as
- several ICH guidelines on the preparation of a well-structured Common Technical Document (CTD).
The first Annex XVI certificate was issued in December 2022 by TÜV SÜD Product Service GmbH for a class III hyaluronic acid-based dermal filler.10 Considering the complex transitional provisions11,12 with fast-approaching deadlines in 2027 and 2028, the impact on manufacturers (for example on those with dual products, i.e., with both medical and non-medical purposes) may be significant. Only those products that will have a CE mark under the EU MDR will remain accessible on the EU market.
This article outlines some key considerations for manufacturers of Annex XVI products, which are also combination products since they contain an ancillary medicinal substance, such as a hyaluronic acid-based dermal filler that contains 0.3% lidocaine HCL as an ancillary medicinal substance.
EU MDR Scope, Common Specifications, And Directive 2001/83/EC
EU MDR
The scope expansion to products without an intended medical purpose (Article 1 points 2 and 3) reflects the growing concerns about the risks associated with some aesthetic and cosmetic products that function similarly to medical devices. It highlights a few key principles for all stakeholders, such as:
- Learning from medical devices: Even if a product is solely intended for aesthetic reasons, manufacturers and notified bodies must consider the same state-of-the-art practices and harmonized standards used for analogous products that are medical devices (analogous devices as defined under Annex I (2.3) of the CS).
- Clinical investigations: Any clinical investigations conducted in the EU involving products without an intended medical purpose must meet the same regulatory and clinical requirements defined for medical devices in the EU MDR.
- Dual-purpose products: Some products have both a medical and a non-medical intended purpose. One example is a dermal filler, which may be used for aesthetic facial augmentation but also for treating facial lipoatrophy linked to HIV. For these products, manufacturers must comply with both sets of requirements – those for medical devices and for products without an intended medical purpose.
Common Specifications (CS)
Marking an important milestone, the Regulation (EU) 2022/2346, which introduces CS for Annex XVI products, came into force in June 2023 and provides the regulation on how risk management, as set out in the general safety and performance requirements under Annex I of the EU MDR, should be applied to these products. This allows the notified bodies to issue certificates for these products in Annex XVI.
For example, Annex IV of the CS sets specific requirements for dermal filler manufacturers to demonstrate how they address:
- inherent risks linked to the product’s main substance, hyaluronic acid and its source, as well as the final material (gel) properties like rheology, its cross-linking degree and biocompatibility
- procedure-related risks, such as the injection site, repeated use, and the force required for administration
- potential harms including product migration, granuloma formation, vascular complications, blindness, and necrosis
- ancillary medicinal substances, where the use of local anesthetics, often added to fillers for patient comfort, must be verified for quality, safety, and effectiveness, in line with pharmaceutical standards under Directive 2001/83/EC
- clear labeling and instructions for use, such as:
- Administration must be limited to healthcare professionals with appropriate training and national accreditation.
- Products must not be used in individuals under 18 years of age.
Overall, these implementing acts clarify how manufacturers should meet key EU MDR obligations for specific product groups.
Directive 2001/83/EC And Borderline Classification
When a product without an intended medical purpose, such as a dermal filler, contains a medicinal substance as an integral part, such as lidocaine HCL, it is classified as a combination product under EU MDR Article 1(8).
In this case, manufacturers must demonstrate that the medicinal substance has an ancillary action relative to the device, ensuring that the product continues to be regulated as a device rather than a medicinal product. The primary mode of action of the product is therefore key:
- If the action of that substance is ancillary and is supported through data, the product would be a device regulated under the EU MDR. However, a consultation (Annex IX 5.2 to the EU MDR) from a medicinal/ competent authority (e.g., EMA) would be required.
MDCG 2022 – 5 explicitly identifies “Soft tissue fillers incorporating local anesthetics” as an example of devices incorporating, as an integral part, an ancillary medicinal product.
If a primary mode of action is pharmacological, the product is regulated as a drug, even if it’s intended purely for cosmetic use. However, the relevant General Safety and Performance Requirements (Annex I to the EU MDR) for the device part need to be fulfilled.
The complexity for borderline products increases when they incorporate animal-derived ingredients, like bovine collagen in dermal fillers. In these cases, manufacturers must also ensure compliance with the relevant parts of the Regulation (EU) No. 722/2012 and Animal By-Products Regulation (EC) No. 1069/2009. These rules ensure the material is safe and traceable to avoid disease transmission (e.g., bovine spongiform encephalopathy).
Regulatory Key Milestones
Qualification And Classification
Qualification and classification are pivotal early-stage milestones. Ideally, the manufacturer should share the outcome with its notified body during an early structured dialogue13,14 to avoid late identification of roadblocks or disputes in borderline classification, which may have a significant impact on the project's success and/or duration.
Manufacturers must qualify and classify their products, ensuring they reference solid, documented evidence to support their claims. In doing so, the following aspects become key in the technical documentation:
- Intended purpose: The product’s intended purpose, including any aesthetic indications, must support its qualification as a product without an intended medical purpose. Additional information is provided in the MDCG 2023-5 guidance on the qualification and classification of Annex XVI products.6
- Mode of action: Scientific evidence must show that the device works primarily via a mechanical/physical mode of action once it is injected into the relevant dermal layer and does not have a principal pharmacological, immunological, or metabolic effect.
- Dual-purpose products: If a product is claimed to have both a medical and a non-medical intended purpose, it must still meet the qualification requirements of a medical device according to Article 2(1) of the EU MDR, and the medical purpose must be supported by relevant clinical evidence.
All medical devices regulated under the EU MDR, including Annex XVI products, must be classified in accordance with the rules set out in Annex VIII of the EU MDR. The product’s risk classification determines the applicable conformity assessment procedure, as well as the type and level of non-clinical and clinical data that manufacturers must provide.
Consultation procedure for combination products in accordance with Annex IX 5.2 to the EU MDR
When a medicinal substance is incorporated into a medical device, the notified body is responsible for verifying the usefulness of the substance as part of the device. During the conformity assessment procedure, the notified body will seek a scientific opinion from one of the competent authorities designated by the Member States in accordance with Directive 2001/83/EC or from the EMA on the quality and safety of the medicinal substance, including the benefit or risk of the incorporation of the substance into the medical device.
It is pivotal to note that the consulted medicinal products authority will consider the manufacturing process and other quality data relating to the usefulness of incorporating the substance into the device. For this purpose, the manufacturer must submit documentation on the medicinal substance itself and as an integral part of the medical device in CTD format.
The EU MDR, in Annex IX, Chapter II, Section 5.2(d), defines the timelines for the consultation with the medicinal products authority, which could prolong the conformity assessment review and certification process by the notified body:
- Once a notified body submits all necessary documentation, the medicinal products authority has up to 210 days to issue its scientific opinion.
- If the manufacturer wants to make any changes to an ancillary substance already incorporated into a device, the notified body is to be informed before implementing the change. The notified body then seeks the opinion of the medicinal products authority, and the authority must provide its opinion within 60 days of receiving all necessary documentation related to the change.
The consulted medicinal products authority may issue an unfavorable scientific opinion, and in most of those cases the notified body would not issue a certificate.
Expert Opinions: Clinical Evaluation Consultation Procedure (CECP)
Dermal fillers classified as a class III implantable device are subject to the Clinical Evaluation Consultation Procedure as per Article 54 of the EU MDR.15 When exceptions under paragraph 2 of Article 54 do not apply, the notified body must submit its clinical evaluation assessment report to the screening and expert panel at the European Commission.
Products without a medical purpose that, for example, include a medicinal substance, like dermal fillers incorporating a local anesthetic, are not just governed under the EU MDR but also need to consider other European legislations. This hybrid nature presents unique challenges for manufacturers in achieving regulatory compliance. They must adopt a proactive, multidisciplinary approach, integrating regulatory strategy, product design and development, and clinical evidence to ensure both market entry and ongoing compliance in the evolving aesthetic product environment.
Key Considerations For Manufacturers
- Engage early with notified bodies and authorities to limit possible challenges during the conformity assessment review.
- Perform comprehensive risk assessments during product development and note that special attention should be given to animal-derived materials and ancillary medicinal substances.
- Develop robust clinical data and a thorough clinical evaluation; the stringent requirements of the EU MDR do not differ for products without a medical purpose.
- Build a broad regulatory intelligence across all relevant regulatory frameworks for your products and monitor the constantly evolving landscape of required standards and guidance documents.
References
- Hong, S., Lee, J.R., & Lim, T. (2010). Pigment deposition of cosmetic contact lenses on the cornea after intense pulsed-light treatment. Korean Journal of Ophthalmology, 24(6), 367–370.
- EMJ Dermatol. 2024;12[1]:63-65. https://doi.org/10.33590/emjdermatol/ZIMR6630.
- LIDOCAINE HYDROCHLORIDE MONOHYDRATE European Pharmacopoeia monograph No. 227 a
- Commission Implementing Regulation (EU) 2022/2346 of 1 December 2022 laying down common specifications for the groups of products without an intended medical purpose listed in Annex XVI to Regulation (EU) 2017/745 of the European Parliament and of the Council on medical devices
- Manual on borderline and classification for medical devices under Regulation (EU) 2017/745 on medical devices and Regulation (EU) 2017/746 on in vitro diagnostic medical devices Version 4 – September 2025
- MDCG 2023-5 Guidance on qualification and classification of Annex XVI products A guide for manufacturers and notified bodies December 2023
- MDCG 2020-12 Guidance on transitional provisions for consultations of authorities on devices incorporating a substance which may be considered a medicinal product and which has action ancillary to that of the device, as well as on devices manufactured using TSE susceptible animal tissues. June 2020
- MDCG 2022 – 5 Rev. 1 Guidance on borderline between medical devices and medicinal products under Regulation (EU) 2017/745 on medical devices October 2024
- Volume 2B Notice to Applicants Medicinal products for human use Presentation and format of the dossier Common Technical Document (CTD)- (May 2008).
- Medical Device Regulation (MDR) 9 November 2023 TÜV SÜD releases the worldwide first certificate for aesthetic products according to Annex XVI of the MDR Munich. The Notified Body TÜV SÜD Product Service GmbH released the first certificate for a non-medical purpose device according to Annex XVI of Regulation (EU) 2017/745. The certified device – a hyaluronic acid based dermal filler – is covered by group 3 of Annex XVI and fulfils the strict requirements of MDR and the Common Specifications 2022/2346.
- Regulation (EU) 2023/1194 of 20 June 2023 amending Implementing Regulation (EU) 2022/2346 as regards the transitional provisions for certain products without an intended medical purpose listed in Annex XVI to Regulation (EU) 2017/745 of the European Parliament and of the Council
- Regulation (EU) 2023/1194 of 20 June 2023 amending Implementing Regulation (EU) 2022/2346 as regards the transitional provisions for certain products without an intended medical purpose listed in Annex XVI to Regulation (EU) 2017/745 of the European Parliament and of the Council
- MDCG 2022-14 MDCG Position Paper Transition to the MDR and IVDR Notified body capacity and availability of medical devices and IVDs August 2022 (action 15).
- MDCG 2019-6 Rev5 Questions and answers: Requirements relating to notified bodies Revision 5 - February 2025
- M. Kebede, M. Fink. Key Takeaways From The EU MDR Expert Panels' First Published Scientific Opinions. Clinical Leader Guest Column | July 10, 2023. https://www.clinicalleader.com/doc/key-takeaways-from-the-eu-mdr-expert-panels-first-published-scientific-opinions-0001
About The Authors:
 Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
 Matthias Fink, M.D., is a senior managing consultant at AKRA Team Inc. He advises medical device and in vitro diagnostic manufacturers, from startups to global leaders, on regulatory and clinical strategies, with a focus on EU MDR and U.S. FDA requirements. He is a former clinical reviewer and manager of the Clinical Focus Team North America at TÜV SÜD Product Service, one of Europe’s largest notified bodies, with experience in both Germany and the U.S. He is also a Board-certified orthopedic and trauma surgeon with 17 years of clinical experience in orthopedic, trauma, and reconstructive surgery, as well as training in cardiovascular and thoracic surgery. Fink is a frequent speaker at national and international conferences and workshops, specializing in clinical evidence generation and regulatory requirements and strategies.
Matthias Fink, M.D., is a senior managing consultant at AKRA Team Inc. He advises medical device and in vitro diagnostic manufacturers, from startups to global leaders, on regulatory and clinical strategies, with a focus on EU MDR and U.S. FDA requirements. He is a former clinical reviewer and manager of the Clinical Focus Team North America at TÜV SÜD Product Service, one of Europe’s largest notified bodies, with experience in both Germany and the U.S. He is also a Board-certified orthopedic and trauma surgeon with 17 years of clinical experience in orthopedic, trauma, and reconstructive surgery, as well as training in cardiovascular and thoracic surgery. Fink is a frequent speaker at national and international conferences and workshops, specializing in clinical evidence generation and regulatory requirements and strategies.