
EU Medtech Labeling Compliance & The Competitive Advantage Of A Digitalized Process
By Rowan Nidd and Maggie Chan, PA Consulting

Optimizing and digitalizing labels can provide the medtech industry with a significant competitive advantage by enhancing speed to market, ensuring products remain on the market, reducing costs, and alleviating resource burdens. In an industry where time and regulatory compliance are crucial, efficient labeling processes are essential for bringing medical devices to market quickly and play a vital role in keeping products available to patients and healthcare providers. According to MedTech Europe’s survey on the availability of medical devices in 2022 in connection to Medical Device Regulation (MDR) implementation, there is an average lead time of 13 to 18 months for a labeling change to be fully implemented.
Below we examine the key challenges within labeling that are playing into these long lead times and how labeling optimization and digitalization could reduce lead times and costs of placing – and maintaining – a device on the market.
Labeling Requirements In The EU
Under the EU MDR 2017/745 Annex I Chapter III, each medical device is required to be accompanied by information needed to identify its manufacturer and its intended use for users and other individuals to distinguish from similar devices and ensure the safety and performance of the device.
There are several components of labeling for medical devices, as listed below, but ultimately the intention of labels is to ensure patients and/or relevant users have access to the intended use, warnings, risks, and all potential adverse events. More importantly, labels provide traceability, which allows manufacturers and/or healthcare professionals to track the device’s users when a communication on a safety issue is required (i.e., recall, newly identified risks, updated precaution, etc.).
- Device Labeling: These are labels placed on the device itself, providing information about the device and manufacturer with UDI and symbols that indicate the storage/handling requirements, warnings/precautions, etc.
- Package Labeling: These are labels on the device package, including cartons and sterile packaging, with device information, storage/handling requirements, etc.
- Instructions for Use (IFU): This is a document that includes intended use, warnings, and procedural/operating instruction, and other relevant safety and performance information
- Product Information for Patient (PIP): This is a document that is required to be provided to patients with an implantable device. This document summarizes the intended use/indication, risks/precaution, and other relevant information that the patients need to know when taking care of the device.
- Implant Card: This document is required to be provided to patients with an implantable device. This card includes device information and manufacturing information required for identification purposes.
- Summary of Safety and Clinical Performance (SSCP): This document is required for class IIa, IIb, III, and implantable devices; it provides information about the safety and clinical performance of the medical device to the patient and public under the EU MDR 2017/745. The document will be available through Eudamed, the IT system developed by the European Commission to implement EU MDR on medical devices.
Functional Team Responsibilities For Improved Labeling Compliance
Compliance with labeling regulations across different markets requires cross-functional expertise, which can have a significant impact on your organization. To ensure all labeling components are compliant with the relevant regulations, a cross-functional capability that includes regulatory, R&D, quality, clinical, manufacturing, and marketing is needed. It is essential for organizations to build a capability that can assess the regulatory landscape, taking into account the current and future product portfolio. Ideally, this capability will be responsible for conducting regulatory assessments, providing guidance to the project team, and establishing trusted relationships with regulators. Labeling regulations are complex and vary across markets (labeling format and content), languages, and channels (paper vs. electronic) and are regulated differently based on the risk level of the device and the targeted market, so an internal capability with input from regulators is essential to optimize the approval process. This capability usually sits within the existing regulatory function, as the responsibilities are usually aligned with the primary functions of the regulatory team and will serve as gatekeepers to ensure all labeling components meet the regulation requirements prior to submission.
After the labeling content is approved by the internal teams and regulatory agencies, a series of downstream activities happen. These include manufacturing, distribution, marketing, customer support, and supply chain, which are responsible for updating and distributing the updated labeling content to users, including healthcare professionals, users, patients, and other impacted individuals. The table below summarizes the key additional responsibilities needed by teams to comply with labeling regulations.
Table 1: Summary of the key additional responsibilities for teams to comply with labeling regulations. Please note, responsibilities are varied based on the local regulations.
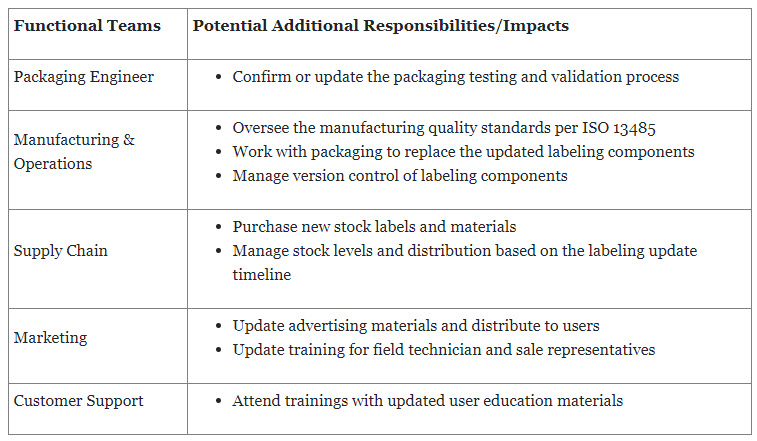
In summary, labeling regulations have wide-ranging organizational impacts. These impacts affect regulatory, R&D, manufacturing, distribution, supply chain, and marketing functions within the organization. Understanding and addressing these impacts are essential to ensure compliance, maintain operational efficiency, and effectively manage the labeling process throughout the organization.
External & Internal Triggers For Label Updates
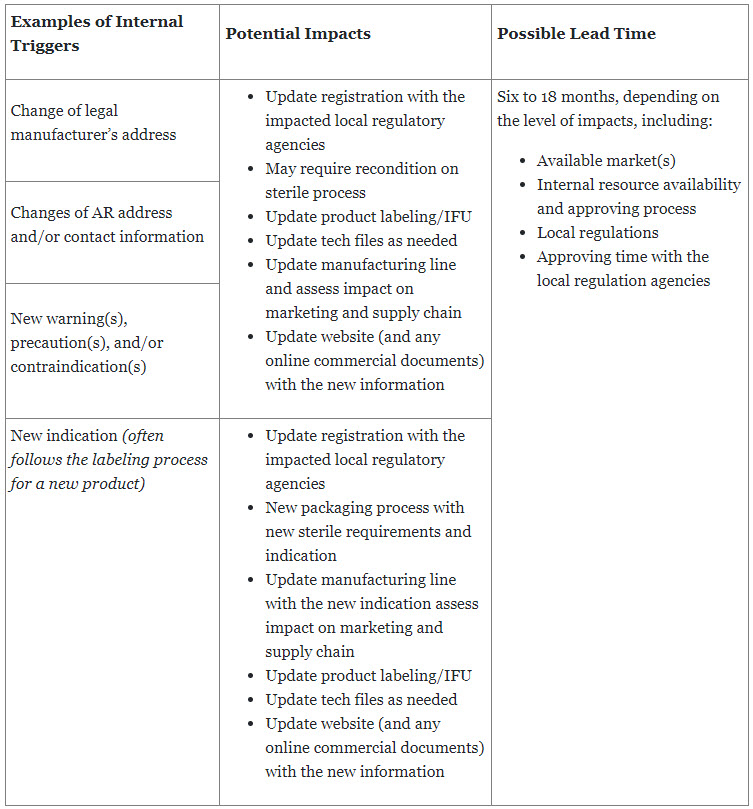
Manufacturers are required to provide the latest safety and performance information of a device to patients and users to ensure the safety, quality, and efficiency of the device as intended. This also continues for the entire life of the device once it is placed on the market or put in service. Updates to labeling can be triggered by both external and internal elements, which requires that manufacturers establish a standardized process to monitor all potential triggers while minimizing the impact to the business.
Table 2: Examples of external triggers with possible average lead time
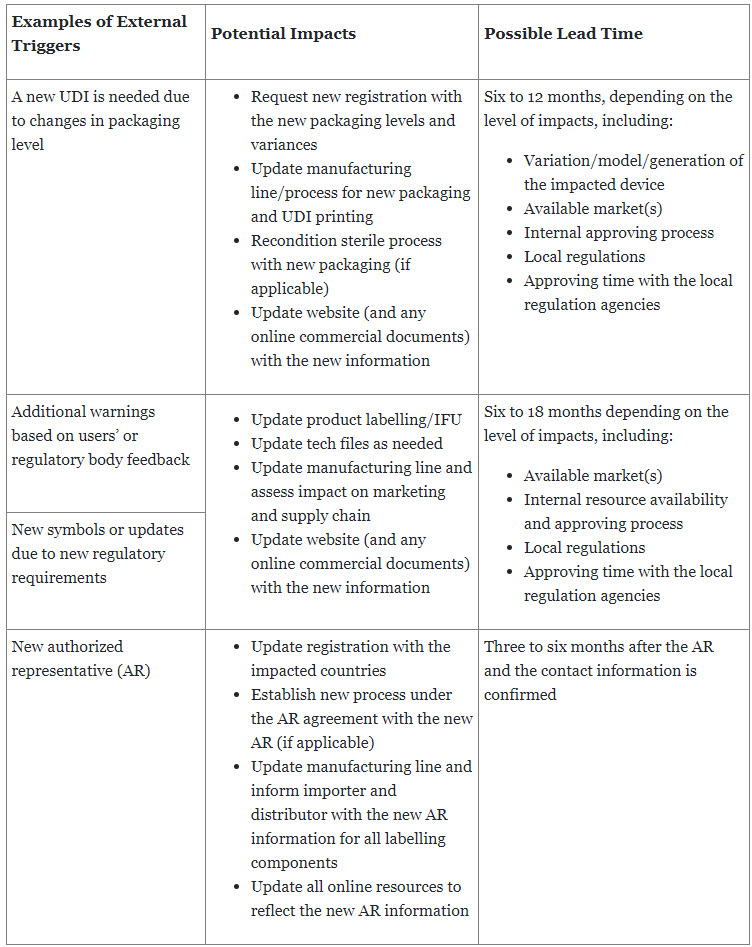
Table 3: Examples of internal triggers with possible average lead time
Benefits Of An Optimized And Digitalized Labeling Process
The major benefits of optimizing and digitalizing the labeling process are increased speed to market by minimizing approval time, reduced development and maintenance costs on labeling, and standardized ways of working with defined roles and responsibilities for all stakeholders within the labeling project team. A centralized system provides transparency throughout the end-to-end labeling process, allowing manufacturers to manage labeling content and expedite the approval process. This not only prevents redundancies but also reduces the risk of errors and inconsistencies across different markets.
By streamlining the labeling process, manufacturers gain control of the entire process, from content development to supply chain, which reduces the risks of non-compliance and product recalls. Standardized labeling processes with defined roles and responsibilities enable manufacturers to provide adequate training to specific users and maintain quality control. An optimized labeling process ensures accurate, complete, and compliant labeling submissions, thereby reducing the number of review iterations and expediting the approval process. Consistency in labeling submissions provides evidence to Notified Bodies that your company understands the regulations and has the capability to adapt to updates and stay in compliance, ultimately shortening the time to market for medical devices – and as a result, gaining advantage over competitors’ products.
Implementing a centralized system reduces the risk of errors resulting from manual workarounds and accelerates the labeling process. This leads to cost savings by reducing resource requirements and decreasing the overall time spent. It also eliminates the need to invest in additional regulatory resources to fill knowledge gaps. Additionally, a centralized system reduces the risk of non-compliance by automatically assessing the potential impact on all relevant labeling components. Non-compliance with labeling regulations can have severe consequences, including regulatory penalties, product recalls, and reputational damage. By implementing robust labeling processes, organizations can mitigate the risk of non-compliance. This includes ensuring accurate and up-to-date label content, proper translations, adherence to regional requirements, and maintaining effective change control mechanisms. By reducing the risk of non-compliance, organizations can avoid costly product recalls and protect their market presence and reputation.
Conclusion
Optimizing and digitalizing the labeling process will bring significant advantages to medical device manufacturers by reducing time to market through streamlined processes, minimizing resource burden and costs, eliminating duplication of effort, and mitigating the risk of non-compliance. Organizations can achieve greater operational efficiency, cost savings, and improved market competitiveness. Investing in an optimized labeling process is a strategic approach that can yield significant returns in terms of faster time to market, reduced regulatory costs, and enhanced compliance.
About the Authors:
 Rowan Nidd is a regulatory affairs and labeling expert at PA Consulting. He specializes in process improvement, operational excellence, and digitization to increase speed to market for new products and to keep products on the market. He leads large regulatory, labeling, and digital transformation projects that include using structured content authoring and AI/machine learning to automate document and systems population for automation, speed, and efficiency. He has an MSc and BSc in molecular genetics from the University of Copenhagen, Denmark.
Rowan Nidd is a regulatory affairs and labeling expert at PA Consulting. He specializes in process improvement, operational excellence, and digitization to increase speed to market for new products and to keep products on the market. He leads large regulatory, labeling, and digital transformation projects that include using structured content authoring and AI/machine learning to automate document and systems population for automation, speed, and efficiency. He has an MSc and BSc in molecular genetics from the University of Copenhagen, Denmark.
 Maggie Chan is a life sciences and regulatory expert at PA Consulting. She focuses on leading process and operation improvement for medical device and pharmaceutical companies. She has led labeling remediation and e-labeling process design projects in compliance with global medical devices regulation for both implantable and non-implantable medical devices. She has been supporting companies by building capabilities and designing processes to ensure they comply with ISO 13485, EU MDR, 21 CFR, etc., in line with the product portfolio. She has a Master of Science in law from Northwestern Pritzker School of Law in the U.S., and a BS in biology.
Maggie Chan is a life sciences and regulatory expert at PA Consulting. She focuses on leading process and operation improvement for medical device and pharmaceutical companies. She has led labeling remediation and e-labeling process design projects in compliance with global medical devices regulation for both implantable and non-implantable medical devices. She has been supporting companies by building capabilities and designing processes to ensure they comply with ISO 13485, EU MDR, 21 CFR, etc., in line with the product portfolio. She has a Master of Science in law from Northwestern Pritzker School of Law in the U.S., and a BS in biology.