
FDA Inspections And UDI — What To Expect And How To Prepare
By Dan O'Leary, President, Ombu Enterprises

The FDA has not provided specific information about unique device identification (UDI) related activities as part of inspections. In particular, the agency has not updated the Quality System Inspection Technique (QSIT) or issued a Compliance Program Guidance Manual (CPGM). However, it is clear that inspections will include UDI related activities, since they are now part of the regulatory requirements.
In a recent webinar, my colleague John Avellanet of Cerulean Associates reported that, starting in the fourth quarter of 2015, FDA would issue Warning Letters to those firms with little to no progress in UDI compliance. His expectation, because of the preparation time for Warning Letters, implies third quarter inspections would include UDI and generate a Form 483. This is consistent with the effective date for the regulations, December 23, 2013, and the first compliance date for Class III devices, September 24, 2014. The compliance date for life sustaining, life supporting, and implantable devices is September 24, 2015.
Prepare For Implementation
Even though FDA has not provided specific direction to investigators, it is straightforward to infer how an investigator would prepare for an inspection. In addition, the FDA’s Global Unique Device Identification Database (GUDID) has the potential to provide additional insight for an investigator.
As a device manufacturer, you do not need to wait for an FDA inspection. Because you have access to the information, be sure to include these points in your internal quality audit program.
Step 1 – Get the regulations.
The UDI revisions were effective as of December 23, 2013, so you should have them readily available and implemented. The compliance dates, relating to specific devices, are on a different schedule.
Table 1 lists the parts of the FDA regulations affected by the UDI rule. Download them and incorporate them in your QMS; they are documents of external origin. Some parts may not apply to every device manufacturer. For example, 21 CFR Part 821 - Medical Device Tracking applies to a limited number of companies. On the other hand, Part 806 - Corrections and Removals applies to all device manufacturers.
Table 1: Parts Affected by the UDI Rule
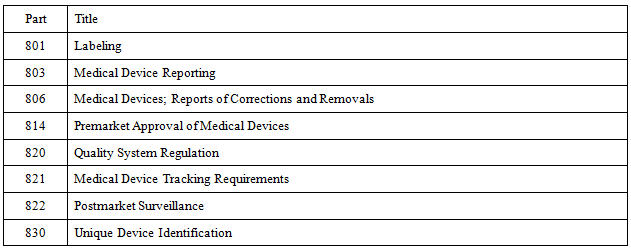
Be cautious with Part 803 - Medical Device Reporting. There is a change pending with an effective date of August 14, 2015, that implements the eMDR (electronic medical device report) rule. It will change some sections affected by the UDI rule.
Many companies used printed copies of the regulations, especially the QSR (quality system regulation), often in small books that fit in a pocket. Be sure that you have the version that includes UDI. An easy way to check is to look at the definitions. If the copy of QSR, for example, does not include 803.3(cc), which defines unique device identifier (UDI), then you have an obsolete version that you should replace.
Step 2 – Implement the regulations.
After downloading the regulations, identify the explicit changes for UDI. Use your word processor’s find capability to identify all of the places that include “unique device identification”, “UDI”, “device identifier”, “DI”, “production identifier”, or “PI”.
For each of the regulation changes, identify: a) any new procedure or work instruction required, b) any existing procedure or work instruction to modify identified by document number, revision, and section, c) any designated individuals who need new skills, and d) any new records to retain and their record retention period. The UDI record retention period is “3 years from the date the labeler ceases to market the version or model”, but the QSR record retention period is “Design and expected life of the device, but in no case less than 2 years from the date of release for commercial distribution by the manufacturer.”
For example, 820.120(b) adds “correct unique device identifier (UDI)” to the label accuracy check. This might require:
- A revised work instruction to include the UDI check,
- A new work instruction to read the automatic identification and data capture (AIDC) portion of the label, and
- A new skill set for the designated individuals who operate the scanner and determine the UDI is correct.
Of course, there are additional requirements for document change control, job descriptions, and training records.
Step 3 – Establish a compliance date implementation schedule.
Develop a schedule to bring devices into compliance on the specified compliance date. Three dimensions affect the compliance date: device class, device characteristics, and UDI activity. Make a list of every device you ship and classify it along the three dimensions. From the list, you can determine the compliance date and develop an implementation plan.
Device Class
- Class III
- Class II
- Class I
- Unclassified
Device Characteristics (Some devices may be in more than category.)
- Life sustaining
- Life supporting
- Implantable
- Intended to be used more than once and intended to be reprocessed before each use
- Stand-alone software
- Exempt from QSR, Part 820
UDI Activity (Most devices will have more than one category.)
- UDI on the device label
- UDI on the device packaging configuration
- Direct marking on the device
- Load the DI record into GUDID
For example, consider a Class II device that the manufacturer intends to be used more than once and reprocessed before each use. This device has two compliance dates.
By September 24, 2016, the labels and packages must have a UDI, the dates on the labels have the correct format, and GUDID has the device information.
By September 24, 2018, the device must have a UDI as a permanent marking.
Step 4 – Present the project at management review.
ISO 13485:2003 requires “new or revised regulatory requirements” as a management review input. While it is not required in QSR, most device companies include it in their management review procedure. Be sure to document the agenda item and record the participants.
Prepare For Inspection
The FDA investigator will prepare for the inspection by checking information that FDA holds — information that you provided. The investigator will match this information with your procedures and records during the inspection.
Before the inspection, the investigator might do the following:
- Retrieve a sample of the devices listed including the device class, product code, and premarket submission number.
- For the items in the sample, retrieve the device identifiers, the issuing agency, and the packaging configurations from the GUDID DI record for each premarket submission number.
- For the items in the sample, retrieve any MDRs the firm submitted since the UDI rule went into effect.
- For the items in the sample, retrieve any correction and removal reports the firm submitted since the UDI rule went into effect.
With this information in hand, it is easy for the investigator to check procedures and records.
Retrieve a sample of procedures and work instructions that implement UDI.
- Verify that the changes correctly implement the regulation.
- Verify the review and approval of the changes following 820.40 - Document Control.
- Verify training records of the employees affected by the changes.
Review the device identifiers previously retrieved from GUDID.
- Verify that the firm documented the limits of specifications, performance, size, and composition that define the version or model represented by the device identifier.
- If there is more than one DI for the same premarket submission number, determine the reason, i.e., the device comes in multiple sizes.
- If there is a new DI created because of a change, verify the design change records following 820.30(i).
- If there is a new DI created because of a change, verify evaluation of the change against the criteria for a new 510(k). Hint: Check the DHF for the records in the 510(k) analysis guidance document.
Review the issuing agencies previously retrieved from GUDID.
- Verify that the firm has established and maintains records of the issuing agencies as an acceptable supplier.
- Verify that the firm has established and maintains records of the GMDN agency as an acceptable supplier.
- Determine if the firm uses a third party to help maintain GUDID. If so, verify the firm established requirements, evaluated the potential supplier, and maintains records as an acceptable supplier.
Review the device identifiers previously retrieved from GUDID.
- Verify that the labeling inspection checks the accuracy of the UDI.
- Verify the job description and training records of the designated individual who performs the labeling inspection.
- Check some labels in current production to verify the UDI information in both the easily readable plain text and the AIDC.
- Check some labels in current production to verify the correct date format.
- Check some labels in current production to verify the AIDC follows the issuing agency specifications.
Review the device identifiers previously retrieved from GUDID.
- Verify that the primary label recorded in the device history record (DHR) includes the required UDI elements.
- Verify that the DHR includes the UDI.
- Verify the job description and training records of the designated individual who authorizes release after DHR review.
Review the device identifiers previously retrieved from GUDID.
- Sample products in stock released for distribution to verify the DI on individual device labels and packaging configurations.
- Obtain a sample of distribution records and verify that each packaging configuration has a device identifier in the GUDID DI record.
Review the device identifiers previously retrieved from GUDID.
- Review complaint records to verify the record includes the UDI.
- Review service records to verify the record includes the UDI.
Review any MDRs the firm submitted since the UDI rule went into effect.
- Review the MDRs to verify each one includes the UDI or an explanation of why the firm didn’t provide it.
- For MDRs with a UDI, verify that the associated complaint record has the same UDI.
Review any correction and removal (C&R) reports the firm submitted since the UDI rule went into effect.
- Verify that the C&R report has a correct UDI based on the associated records of the actions.
- For any C&R initiated but not reportable, verify that the C&R record has a correct UDI based on the associated records of the actions.