
FDA's Safety And Performance-based Pathway: An Alternative To Substantial Equivalence For 510(k) Submissions
By Jeffrey S. Eberhard, Freyr

Since the inception of the 510(k) program, the 510(k) clearance of medical devices has been based on their proven substantial equivalence with claimed predicate device(s). In concert with the goal of adopting the least burdensome approaches, the FDA provides an alternate pathway based on proven safety and performance characteristics, instead of devices’ equivalence to other predicates. This pathway is an expansion of the abbreviated 510(k) pathway, applicable to some well understood low to moderate-risk class II device categories. The FDA has released and continues to release device-specific guidelines to encourage manufacturers to opt for this approach for their device approvals. The FDA also conducts webinars and workshops to assist industry stakeholders understand the pathway.
The pathway is voluntary and is not mandated by the FDA. Though the manufacturer is not required to prove substantial equivalence of its device with a predicate device, the manufacturer is still required to identify a predicate device in the scope of the submission. Manufacturers can opt for the safety and performance-based pathway if the device has the same indications for use as the identified predicate, its technological characteristics do not raise any different safety and effectiveness concerns than the identified predicate, and it meets all the FDA-identified performance criteria for the given device. If any of the above factors are not met, the manufacturer can opt to submit a traditional, special, or abbreviated 510(k).
The FDA has so far identified performance and safety criteria and testing methodologies for spinal plating systems, orthopedic non-spinal metallic bone screws and washers, magnetic resonance receive-only coils, cutaneous electrodes for recording purposes, and conventional foley catheters, and the final guidance is in effect for each. The draft guidance for soft (hydrophilic) daily wear contact lenses has been released, with the final guidance not yet available. For each type of device, the guidance includes the description of the device, the types of devices included and excluded under the purview of the safety and performance-based pathway, applicable performance criteria that are to be met by the device, and the recommended testing methodologies. A brief outline of these device categories is detailed in the table below.
Table 1: Safety and Performance-based Pathway Device Categories
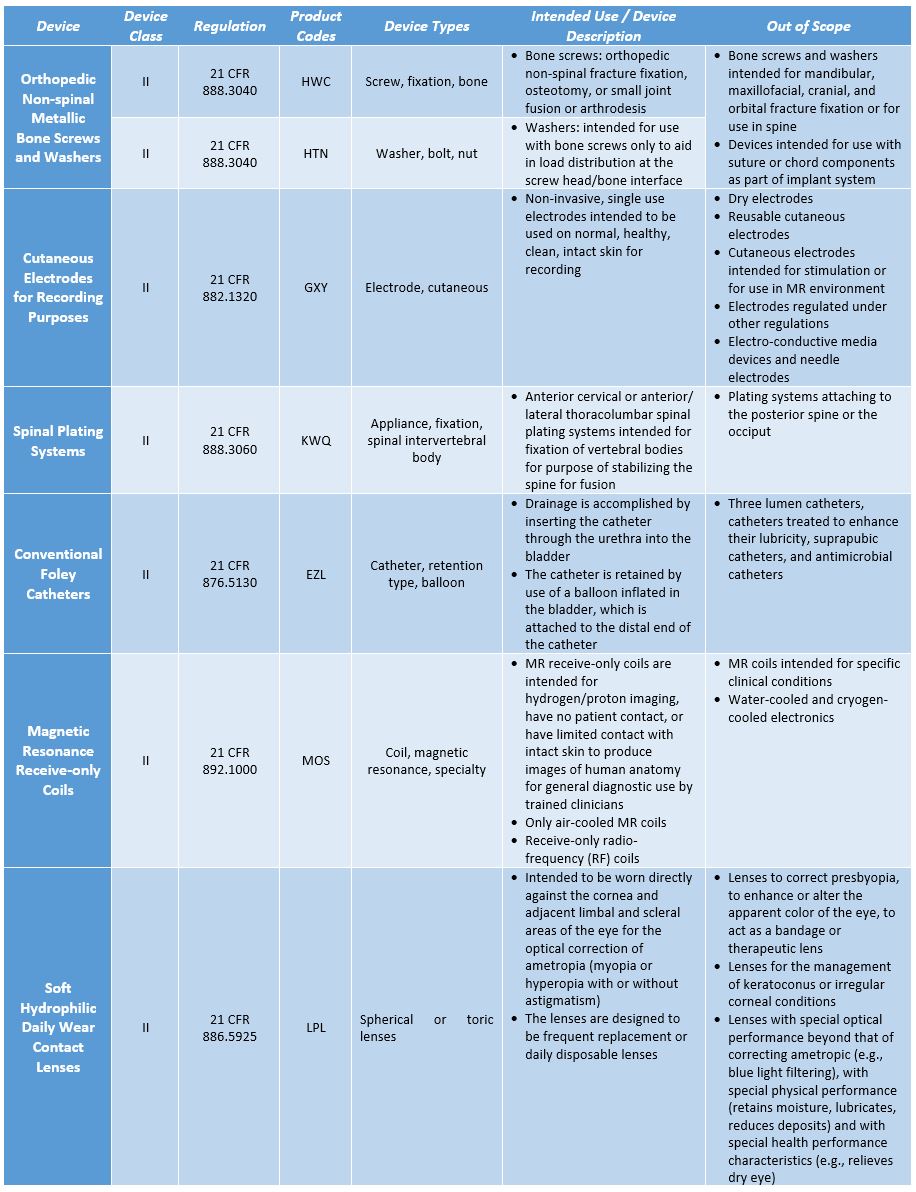
The performance criteria defined in these guidelines ensure that the new device is at the least equivalent to legally marketed devices, in terms of safety and performance. The safety and performance can be demonstrated based on the FDA’s recognized consensus standards, the FDA guidance, special controls, scientific literature, or submission of historical data. While opting for this pathway, the manufacturer should not use performance criteria suggested in standards that are not recognized by the FDA. Some tests would require complete test protocols and all test reports and the summary of test results and declaration of conformity would be sufficient for submission, as a part of 510(k) application.
When the performance criteria are included in the FDA recognized consensus standard and the manufacturer uses the same testing methodology included in the FDA recognized consensus standard, submitting a declaration of conformity would suffice under this pathway. When the performance criteria are established by the FDA in the safety and performance guidance for a given device category and the test methodology from the FDA recognized standard is adopted by the manufacturer, a summary of results should accompany the declaration of conformity. In cases where the performance criteria are established by the FDA in the safety and performance guidance for a given device category and the test methodology is recommended or specified by the FDA, a testing protocol is required. If the test methodology is neither included in the recognized standard nor recommended by the FDA, or if the manufacturer uses its in-house test method as an alternative, the manufacturer shall submit the complete test report. However, manufacturers should note that the FDA does not consider performance criteria that are not included in the device-specific safety and performance-based guidelines.
The table below shows the data that should be included in the submission under various possible scenarios.
Table 2: Data required under various possible scenarios.
|
Type of Performance Criteria and Methodology the FDA identified for Safety and Performance-based Pathway |
||
|
Performance Criteria |
Testing Methodology |
Safety and Performance-based Pathway 510(k) Submission Should Include |
|
FDA-recognized standard |
FDA-recognized standard |
Declaration of Conformity |
|
FDA-established |
FDA-recognized standard |
Results Summary and Declaration of Conformity |
|
FDA-established |
FDA-recommended or specified |
Results Summary and Testing Protocol |
|
FDA-established |
None specified/recommended or alternative to the FDA-specified methodology used |
Complete Test Report |
The submission process, cover letter, Refuse To Accept (RTA) checklist requirements, the review process, e-copy requirements, and MDUFA fees remain the same as for other types of Pre-Market Notification pathways like traditional 510(k), abbreviated 510(k), and special 510(k). The timeline for the FDA to review and make a decision on a 510(k) submitted under the safety and performance-based pathway is 90 FDA days.
To comply with the RTA policy guidance, the manufacturer shall include the sections listed below in the same order. Where a particular section is not applicable for a given device category, the manufacturer can retain the section heading and include the statement, “This section does not apply” or “N/A” for ease of review by the FDA staff. The statement should provide the rationale for why a particular section is not applicable for the device.
Table 3: Required sections for safety and performance-based pathway.
|
|
The manufacturer shall demonstrate the device’s compliance to a standard through the declaration of conformity to the standard, results summary, or a summary report, if recommended in any relevant device-specific guidance, testing protocols, and/or a complete test report demonstrating that the new device meets the FDA-identified performance criteria. The manufacturer shall identify a predicate and provide a trade name, model number, name of the 510(k) submitter/holder, and 510(k) number, if available. Though the safety and performance-based pathway does not require the manufacturer to compare performance specification testing with a predicate device, the manufacturer shall provide a comparison with predicate device in terms of indications for use and technology. For other sections of the 510(k) technical file, i.e., Proposed labeling, sterilization and shelf life, biocompatibility, software, electromagnetic compatibility and electrical safety, and performance testing, the data shall be submitted in terms similar to a typical 510(k) technical file, though it is not a direct comparison with the predicate device.
Below is an example of the test methodologies, performance criteria, and data submission requirements defined for MR coils.
Table 4: MR coil requirements as per the safety and performance-based pathway.
|
Test |
Test Methodology |
Submission Requirement |
Performance Criteria |
|
Image Signal to Noise |
|
Summary of results and Declaration of Conformity |
|
|
Image Conformity |
|
Summary of results and Declaration of Conformity |
|
|
Surface Heating |
|
Summary of results and Declaration of Conformity |
|
|
Acquired Image Quality |
|
Statement from a U.S. Board Certified or international equivalent qualified physician |
|
|
Decoupling Circuit |
|
Circuit diagrams and description of decoupling mechanism |
|
|
Immunity, electrostatic discharge |
|
Summary of results and Declaration of Conformity |
|
|
General electrical / mechanical safety |
|
Summary of results and Declaration of Conformity |
|
The safety and performance-based pathway offers a cost-efficient way for device manufacturers to gain market access in the U.S., as the number of samples that are required to be tested are reduced by half. The FDA is expected to issue a draft and final guidance(s) for additional device types that qualify for the safety and performance-based pathway.
About The Author:
 Jeffrey S. Eberhard is with Freyr and has more than 15 years of experience in the medical device industry serving many key roles in regulatory affairs, quality management systems (QMS), and risk assessment and mitigation. He has led various U.S. device approval projects, including pre-market notifications (510(k)). Jeffrey has developed ISO 13485 and safety testing programs and designed programs for safety, efficacy, and clinical research. Jeffrey has vast expertise in sterile medical devices as well as dermal and oral care medical devices.
Jeffrey S. Eberhard is with Freyr and has more than 15 years of experience in the medical device industry serving many key roles in regulatory affairs, quality management systems (QMS), and risk assessment and mitigation. He has led various U.S. device approval projects, including pre-market notifications (510(k)). Jeffrey has developed ISO 13485 and safety testing programs and designed programs for safety, efficacy, and clinical research. Jeffrey has vast expertise in sterile medical devices as well as dermal and oral care medical devices.