
FDA Updates Draft Guidance On 510(k) Third Party Review Program

By Jof Enriquez,
Follow me on Twitter @jofenriq

The U.S. Food and Drug Administration (FDA) has updated draft guidance on its 510(k) Third Party (TP) Review Program to incorporate elements and requirements set forth under the Medical Device Single Audit Program (MDSAP) by the International Medical Device Regulators Forum (IMDRF).
Section 523 of the Federal Food, Drug, and Cosmetic (FD&C) Act authorizes FDA to accredit third parties to review premarket notification (510(k)) submissions and recommend the initial classification of certain low-to-moderate risk devices. FDA released draft guidance on the TP Review Program in 2013 and is now updating it for the program to be consistent with global regulatory harmonization efforts started by IMDRF in 2011 and fleshed-out in MDSAP documents finalized between 2013 and 2015.
Under the TP Review Program (Figure 1), authorized TP Review Organizations conduct the equivalent of an FDA premarket review of a 510(k) submission and then forward their reviews, recommendations, and 510(k) submissions to FDA for a decision concerning the substantial equivalence of a device. FDA has 30 days to make a determination upon receipt.

Figure 1
The TP Review Program enables FDA “to focus its internal scientific review resources on higher-risk and complex devices, while maintaining a high degree of confidence in the review of low-to-moderate risk and less complex devices by TP Review Organizations, and to provide manufacturers of eligible devices a voluntary alternative review process that may yield more rapid 510(k) decisions from FDA.”
The updated draft guidance contains FDA’s current thinking on four areas:
1. TP Review Organizations’ review of 510(k) submissions (Figure 2)
2. Requirements and recommendations for recognition and re-recognition of TP review
3. Content and format of a TP Review Organization’s application for initial recognition and re-recognition
4. Suspension or withdrawal of recognition
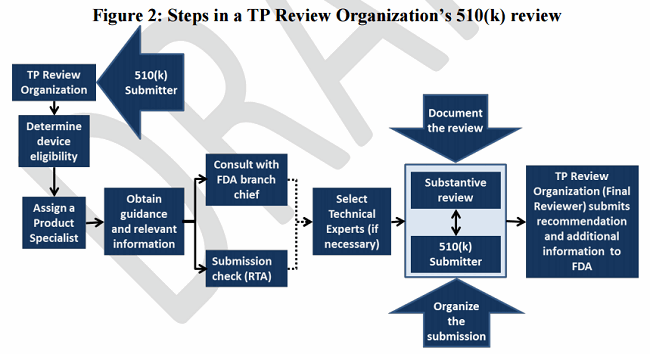
Figure 2
FDA’s new draft guidance draws from four IMDRF MDSAP documents that serve as common criteria for the recognition and monitoring of entities that perform regulatory audits used in multiple jurisdictions, but it is also consistent with the FD&C Act and current FDA regulations.
Related, FDA says IMDRF is in the process of developing a document entitled Competency, Training, and Conduct Requirements for Regulatory Reviewers, which will provide a common set of requirements for personnel involved in reviewing activities.
Due for full implementation in January 1, 2017, the MDSAP program enables device manufacturers to contract with an authorized Auditing Organization (AO) — which currently includes BSI Healthcare and Tuv Sud America — to conduct a single audit against the relevant medical device regulatory requirements of all fully participating regulatory authorities. MDSAP members include Australia’s Therapeutic Goods Administration (TGA), Brazil’s National Health Surveillance Agency (ANVlSA), Japan’s Ministry of Health, Labour and Welfare (MHLW) and Pharmaceuticals and Medical Devices Agency (PMDA), and Health Canada, according to RAPS.