
How The FDA's Pre-Certification Pilot Program Is Paving The Way For AI In Medtech
By Brian Scogland, Halloran Consulting Group

The buzz surrounding artificial intelligence (AI) and machine learning (ML) in the medical technology industry may rival the hype surrounding “big data” from years back. Still, it is undeniable that these technologies already have a place in medtech, and in the future of healthcare.
The many ML products and algorithms cleared by FDA for early detection of health events exist as clear evidence of this fact. These products are on the market and are actively used by physicians; they include offerings by Viz.ai (early stroke detection), Verily Life Sciences (irregular heart rhythm),and AliveCor (atrial fibrillation) — to name just a few.
The FDA, recognizing the growth in this technology area, has launched a Software Precertification (Pre-Cert) Pilot Program to define a new regulatory pathway for companies that develop these software-based medical devices, known as software as a medical device (SaMD). The basic premise is to move away from the traditional 510(k) and De Novo pathways, and to develop a program better suited for technologies that have a rapid development and update lifecycle.
The intent is to provide more streamlined and efficient regulatory oversight, helping these products to reach market safely and operate effectively while not inhibiting time-to-market speed and innovation. SaMD technologies are the next generation of digital health devices and likely will be a major part of healthcare decision-making going forward.
FDA’s pilot launched in September 2017 with 9 companies selected out of over 100 applicants. This cohort has remained intact, and it appears the FDA will not add to this group of companies through the duration of the pilot. The FDA’s initial step was to create a working model of a new regulatory paradigm. FDA has been refining that model ever since, and will begin testing the model in 2019.
The first reaction I hear from most colleagues is that the Pre-Cert Pilot Program represents one of the FDA’s biggest initiatives in recent memory, a fundamental shift in the way FDA views product review. Appreciation for the agency’s transparency throughout process, from launch through each incremental adjustment to the model, has been nearly universal. Consistent with past initiatives, the FDA has rolled out a “road show” to communicate the change well ahead of any proposed implementation date, and to great effect.
The pressing challenge for many companies making SaMD products is the fact that they are observers on the sideline, and cannot yet take advantage of this new paradigm. The only complaint or request that might be levied against the agency would be to offer a speedier pilot so that others can reap some of the potential benefits. Full implementation of this initiative likely is years away, while the technology it is designed to regulate continues to advance and innovate at a much quicker pace. Organizations are champing at the bit to gain both regulatory clarity and a new path forward for their SaMD products.
For now, those outside the pilot must navigate the traditional regulatory pathways of Class I and Class II SaMD products. Challenges that exist within this traditional paradigm are among the drivers of the pilot. For example, when a manufacturer adjusts its algorithm, will that manufacturer need to file a new submission? The traditional assessment of product changes must continue to take place in this premarket clearance model.
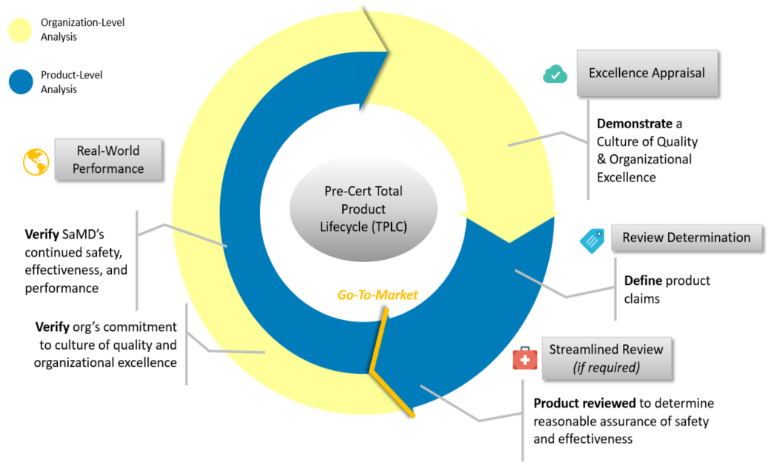
If implemented properly, this new paradigm will benefit SaMD manufacturers by allowing FDA to pre-certify a company by performing an Excellence Appraisal that assesses the company’s “culture of quality” and “organizational excellence” (CQOE), rather than returning to scrutinize every incremental product change. The FDA currently is basing the pilot program's criteria on five Excellence Principles: patient safety, product quality, clinical responsibility, cybersecurity responsibility, and proactive culture.
The most intangible — and potentially hardest — of the five principles for FDA to measure likely is “proactive culture,” wherein an organization is expected to “demonstrate excellence in a proactive approach to surveillance, assessment of user needs, and continuous learning.” This is perhaps the most difficult metric for companies to demonstrate with tangible, objective evidence.
Additionally, the pilot will define precertification based on an organization’s excellence, and then categorize them as Level 1 Pre-Cert or Level 2 Pre-Cert status:
- Level 1 Pre-Cert is designed to allow organizations to develop and market certain lower-risk software without review, while requiring a streamlined review for other types of software. Organizations securing this level of precertification typically will have less experience developing regulated products.
- Level 2 Pre-Cert then allows organizations to develop and market certain lower and moderate-risk software without review, while requiring a streamlined review for other types of software. Organizations securing this level of precertification might include those with experience developing regulated products, as well as those who have implemented mature quality systems and software development processes.
The separation of companies into these two categories should allow the FDA to identify the performance of various types of organizational structures, and to normalize that performance using the Excellence Principles.
While the FDA has not stated as much explicitly in the proposed Working Model for the Pre-Cert Pilot, it should be understood that the backbone of a company’s precertification status will be the FDA’s assessment of the organization’s knowledge and implementation of applicable regulations, international standards, and software development practices (e.g., FDA Quality System Regulation [21 CFR Part 820], ISO 13485, ISO 12207, ISO 62304, ISO 14971, ISO 9001).
The challenge for manufacturers new to this space is to quickly build up expertise in these areas, but exercise patience in allowing themselves the proper amount of practice and runtime to demonstrate excellence in the implementation of these regulations and international standards. The FDA will look to organizations to produce readily-available evidence that they have established a mature software development practice and maintain a constant focus on quality.
Some technologies falling under the Pilot are being developed by relatively young and immature organizations, and these companies likely will need to consider third-party assistance — by organizations or individuals seasoned in regulated industries — to bring their compliance up to speed quickly.
Still, the challenge for FDA is determining how to capture meaningful metrics, indicative of an organization that has proven its focus and commitment to quality, across multiple product types and organization sizes — all while ensuring these companies’ output is both safe and effective.
How can you be involved? Keep your eyes out for user session webinars where the FDA team will share their latest updates. Additionally, FDA is looking for feedback on the latest version of the Pre-Cert working model by March 8, 2019.
About The Author
Brian Scogland, MS, MBA, is Associate Principal Consultant, Quality, with Halloran Consulting Group. Brian’s 10+ years experience in regulatory affairs and quality system development includes creating and implementing quality systems in the medical device industry compliant with ISO 13485 and FDA’s Quality System Regulation. He maintains expertise in software development activities and SaMD. Prior to joining Halloran, Brian was the Regulatory Affairs Manager for lifeIMAGE and held various positions at Boston Scientific and Caliper Life Sciences.
Brian earned his MBA and master’s degree in management from Babson College; he earned his bachelor’s degree in management, with a concentration in Management Information Systems, from Rensselaer Polytechnic Institute.