
How To Stop (Most) Medical Device Recalls Before They Happen
By Marcelo Trevino, independent expert

A device is only as safe and effective as its weakest supplied input. That statement sounds obvious. Yet supplier quality failures remain among the most common root causes of medical device recalls, FDA warning letters, and consent decrees, year after year. The gap between understanding the principle and executing the program is where patient risk lives.
Most medical device manufacturers today source components, subassemblies, raw materials, and services from dozens or even hundreds of external suppliers. A Class III active implant might touch 80 suppliers before it reaches a patient. When something goes wrong in that network, the consequences are the manufacturer's responsibility, regardless of who made the defective part.
This article distills a practical framework for managing supplier quality across the full spectrum: from standard off-the-shelf (OTS) catalog items to fully custom contract-manufactured components. The principles are regulation-grounded, the examples are real-world, and the recommendations are immediately actionable.
Know What You Are Dealing With: The Supplier Spectrum
Not all suppliers carry the same risk profile, and your program cannot treat them as though they do. The first discipline is classification. There are four distinct supplier archetypes in the medical device world, each requiring a different quality posture:
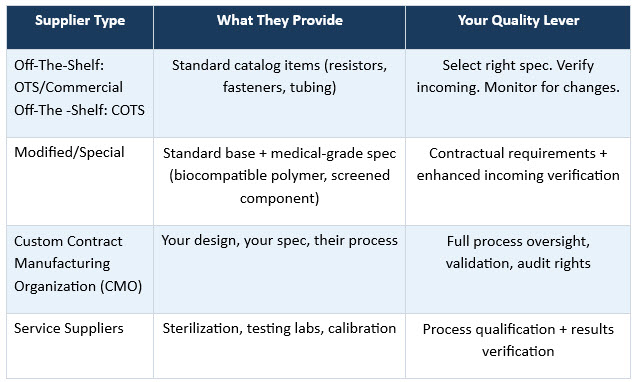
The critical distinction is this: with an OTS supplier, you are selecting from what exists. With a CMO, you own the design, they own the process. That fundamental difference defines every element of how you qualify, contract, and monitor them.
Real-world example: A manufacturer sources a standard transistor for a cardiac monitoring device. The component is OTS; the supplier makes it for dozens of industries. The right quality lever is specification selection (verify it survives device operating conditions), incoming inspection, and a watch on the supplier's product change notifications. Attempting a full ISO 13485 audit of this supplier misallocates resources and creates no real quality benefit. That same audit effort, redirected to the CMO machining your device's titanium enclosure, prevents recalls.
The Regulatory Baseline — And Why It Is More Than A Checklist
Three major frameworks converge on supplier quality requirements: ISO 13485:2016 Section 7.4, the FDA's Quality Management System Regulation (QMSR, effective February 2026), and the EU MDR Annex IX. All three require the same core disciplines: evaluate and select suppliers based on demonstrated ability to meet your requirements; document those requirements formally; verify incoming product; and monitor supplier performance continuously.
The QMSR, the FDA's most significant quality regulation update in decades, now directly incorporates ISO 13485 by reference. For U.S. manufacturers, ISO 13485 compliance is no longer just best practice, it is now federal law.
Where organizations consistently fall short is in executing these requirements with depth. A supplier quality agreement that is a three-paragraph addendum to a purchase order is no quality tool. An annual audit that reviews only the certificate wall and misses the change control log is no risk mitigation. Regulators and third-party auditors see this pattern repeatedly, and it shows up in inspection findings.
Regulatory Quick Reference
- ISO 13485 §7.4: Risk-proportional purchasing controls, supplier evaluation, incoming verification
- FDA QMSR (21 CFR 820, effective February 2026): Now incorporates ISO 13485 by reference — supplier agreements and audit rights are enforceable federal requirements
- EU MDR Annex IX: QMS must address supply chain management; post-market surveillance must trace field failures to supplied components
Common enforcement theme: Inadequate supplier qualification and missing supplier agreements are top-10 FDA 483 observations every year.
Risk-Based Tiering: Apply Effort Where It Matters
The single most powerful structural decision in a supplier quality program is how you allocate intensity. A three-tier model anchored in actual patient risk, not supplier size or relationship history, creates a defensible, efficient, and regulation-aligned program.
Tier your suppliers along two axes: severity (what happens to the patient if this item fails?) and detectability (can you find the failure before it reaches the patient?). A custom-molded fluid path component that contacts sterile saline inside the patient, and whose dimensional failures are not detectable through incoming inspection, belongs at Tier 1. A standard office supply-grade cable tie used only in external packaging belongs at Tier 3.
The link most programs miss: Your design FMEA (DFMEA) already contains this analysis. Every high-severity, low-detectability failure mode in your DFMEA that traces to a supplied component is telling you exactly which supplier deserves Tier 1 treatment. If that link is not formally documented, you have a gap between your design risk analysis and your supplier quality program, and that gap is both a regulatory finding and a patient safety risk.
Formalizing this link requires more than a cross-reference spreadsheet. It demands a living connection: When your DFMEA is updated during a design change, your supplier tier assignments must be reviewed as a mandatory downstream action. Many organizations treat DFMEA updates and supplier qualification as parallel but separate workflows. They are sequential dependencies. A design change that introduces a new failure mode with high severity and low detectability must trigger an immediate reassessment of which suppliers now belong at a higher tier, before that change reaches production.
Real-world example: A device manufacturer had been treating the supplier of a custom silicone membrane as Tier 2, receiving AQL sampling and an annual QMS questionnaire. During a post-market surveillance review, field data showed a clustering of performance complaints correlated with membrane lot variability. Rerunning the DFMEA against the membrane failure mode revealed high severity, moderate frequency, and low detectability once assembled. The supplier was elevated to Tier 1: full on-site audit, process validation review, and skip-lot protocol tied to process performance data. Complaints declined sharply within two production cycles.
Qualification That Holds Up Under Scrutiny
For OTS Components
Qualification is primarily engineering-driven: Verify the component's specification covers your worst-case operating conditions, confirm biocompatibility if applicable per ISO 10993, lock in your approved vendor list to prevent unauthorized substitution, and establish incoming inspection criteria. Critically, understand the supplier's change notification policy before you qualify them. Some OTS suppliers offer proactive product change notices (PCNs). Others do not, meaning the burden of monitoring for changes falls entirely on you.
For Custom CMOs: The Five Non-Negotiables
CMO qualification is process validation under a different roof, not a supplier audit with extra steps. Five elements are non-negotiable:
- Quality System Assessment: Perform an on-site audit for Tier 1 CMOs, not a paper review. Verify ISO 13485 certification, review nonconformance history, and examine CAPA closure rates before you commit to the relationship.
- Process Validation (IQ/OQ/PQ): The CMO's manufacturing process must be validated as rigorously as any internal process. Many manufacturers treat CMO qualification as "we toured the facility and liked what we saw." That is not validation.
- First Article Inspection: This is a full-dimensional, functional, and material characterization of the first production-representative units. For machined implant components, this means coordinate measurement machine (CMM) reports. For molded parts, short-shot studies and material certifications are required.
- Specification Ownership: Your specifications. Your document control system. Your approval authority for any change. If the CMO authored the spec and controls it, you do not control your own product.
- Supplier Quality Agreement: This is a document that specifies change notification triggers in concrete terms: not "material changes" but a specific list: raw material source, tooling, facility, key personnel, sterilization cycle, sub-tier suppliers. Ambiguity here is where recalls are born.
Monitoring: From Scorecards To Supplier-Initiated Changes
Qualification clears a supplier to ship. Monitoring keeps them qualified. Three disciplines carry most of the weight.
Incoming Inspection with Intelligent Sampling
Use AQL-based sampling (ANSI/ASQ Z1.4 for attributes) scaled to the risk tier. For Tier 1 CMOs with non-detectable failure modes, sampling alone is insufficient — process validation data and CMO-generated inspection records should replace or supplement incoming inspection, supported by a formally documented skip-lot protocol triggered by process performance data thresholds. For Tier 3 OTS, a reduced inspection regimen tied to certificate of conformance review is proportionate and defensible.
Performance Scorecards
Share a quarterly scorecard with every Tier 1 and Tier 2 supplier. The metrics that matter most: incoming acceptance rate, CAPA closure timeliness, on-time delivery, and – often overlooked – change notification compliance rate. A supplier with a 99% acceptance rate but a 60% change notification compliance rate is a high-risk supplier that looks low-risk on paper.
The Silent Killer: Supplier-Initiated Changes
This is the single most underestimated risk in supplier quality management. A supplier changes their raw material source, or replaces an aging mold tool, or shifts a process to a lower-cost facility. Each of these decisions can invalidate your design validation, without your knowledge. FDA warning letters and recall investigations surface this pattern repeatedly.
One discipline that reinforces your agreement's specificity requirements is the change log audit. During every Tier 1 audit, request access to the CMO's internal change control records for the trailing 12 months. Review each entry and verify that events meeting your notification triggers were, in fact, communicated. This single practice has more predictive power over supply chain risk than virtually any scorecard metric. A supplier's willingness to open that log, and the quality of what you find inside it, tells you more about the health of the relationship than their acceptance rate ever will.
Real-world example: A device manufacturer experienced a sudden cluster of failures in a polymeric housing component, triggering a field corrective action. Root cause investigation revealed that the CMO had switched resin grades from a supplier that had gone out of stock, using an "equivalent" grade approved internally within the CMO's own material review process. The substitution was compliant within the CMO's QMS, but was never communicated to the manufacturer, whose supplier agreement only required notification of "significant design or process changes." The word "significant" had done most of the damage.
The fix is specificity. Replace vague trigger language in your supplier agreements with an explicit, exhaustive trigger list. Then audit against it, review the CMO's own internal change control log during audits, and verify that notifiable events were actually notified.
Emerging Realities Every Leader Must Prepare For
Software suppliers (SOUP) are now front-line quality risk. Off-the-shelf software components – operating systems, communication stacks, third-party algorithms — cannot be inspected dimensionally. Their "quality" is a function of development life cycle rigor, vulnerability management, and patching commitments. With the FDA's 2023 Cybersecurity Guidance placing post-market software maintenance obligations squarely on device manufacturers, software supplier qualification is no longer optional. If your device incorporates third-party software and you do not have a formal SOUP qualification process, that is a gap your next inspection will find.
The QMSR transition (effective February 2026) tightens sub-tier flow-down requirements. If your supplier agreements do not explicitly require your CMOs and critical OTS suppliers to flow down relevant quality requirements to their own suppliers, update them before your next FDA inspection. Sub-tier visibility is a prerequisite for supply chain integrity. Post-pandemic experience proved that a problem two tiers down can ground your production and jeopardize patients.
Geopolitical concentration risk has become a quality issue. A device whose critical components are single-sourced from a supplier in a geopolitically unstable region is more than a supply chain risk; it is a quality risk if alternative suppliers have not been prequalified and the backup manufacturing process has not been validated. Quality teams are increasingly expected to contribute to supply chain resilience analyses. A prequalified backup supplier with a validated process is a quality asset. Organizations that treat dual sourcing as a quality investment rather than procurement overhead will hold a structural advantage when the next disruption arrives, and it will.
Building A Program That Lasts
The organizations with the strongest supplier quality programs share a common characteristic: They treat supplier quality as a strategic function, not a compliance afterthought. That distinction manifests in three practical ways.
- They invest in supplier capability, not just supplier measurement. Supporting a critical CMO's ISO 13485 certification journey, co-running a process improvement kaizen, or embedding a quality engineer during a new product ramp-up builds the kind of supplier competence that scorecards can only measure, they cannot create it.
- They design their programs to reward supplier transparency. A supplier that self-reports a nonconformance should be handled very differently from one that conceals it. If your corrective action response to a self-reported problem looks identical to your response to a discovered one, you are training your suppliers not to tell you things. That is a cultural failure with direct patient safety consequences.
- They connect supplier quality data to business decisions. Supplier performance metrics belong in management review, alongside internal quality data. Procurement decisions that override quality input — selecting a new CMO for cost reasons without completing qualification or waiving an audit because of schedule pressure — should require formal quality risk acceptance at the executive level. The regulatory requirement for management review of quality system effectiveness is satisfied only when supplier data receives the same scrutiny as internal nonconformance trends, CAPA aging, and audit findings. Anything less is governance in name only.
Quality beyond the factory's walls is a regulatory obligation, a patient safety imperative, and when done well, a genuine competitive advantage. The manufacturers who will thrive in the next decade of tightening global regulation are those who understand that their quality system extends all the way to the last link in their supply chain that could harm a patient.
Apply the tiered framework. Demand specificity in your supplier agreements. Close the loop between your DFMEA and your supplier quality plan. Audit against what matters. And build supplier relationships where quality is a shared goal rather than a contractual obligation that one party enforces and the other endures.
About The Author:
 Marcelo Trevino has more than 25 years of experience in global regulatory affairs, quality, and compliance, serving in senior leadership roles while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, medical imaging/surgical navigation, in vitro diagnostic devices, and medical device sterilization and disinfection products. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR/IVDR, MDSAP). He holds a BS in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. Trevino is also a certified Medical Device Master Auditor and Master Auditor in Quality Management Systems by Exemplar Global. He has experience working on Lean Six Sigma Projects and many quality/regulatory affairs initiatives in the U.S. and around the world, including third-party auditing through Notified Bodies, supplier audits, risk management, process validation, and remediation. He can be reached at marcelotrevino@outlook.com or on LinkedIn.
Marcelo Trevino has more than 25 years of experience in global regulatory affairs, quality, and compliance, serving in senior leadership roles while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, medical imaging/surgical navigation, in vitro diagnostic devices, and medical device sterilization and disinfection products. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR/IVDR, MDSAP). He holds a BS in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. Trevino is also a certified Medical Device Master Auditor and Master Auditor in Quality Management Systems by Exemplar Global. He has experience working on Lean Six Sigma Projects and many quality/regulatory affairs initiatives in the U.S. and around the world, including third-party auditing through Notified Bodies, supplier audits, risk management, process validation, and remediation. He can be reached at marcelotrevino@outlook.com or on LinkedIn.