
Implementing EU MDR and IVDR: Lessons Learned, Part 1
By Marcelo Trevino, independent expert
The transition to the EU’s Medical Device Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR) is not an easy undertaking. As many changes need to be made in the quality system and business processes to meet the new regulations, some organizations are experiencing challenges in implementing and demonstrating compliance to their notified bodies. To start, organizations need to ensure that their devices have the accurate certificate scopes and that current certificates can be maintained until expiration. Several significant important aspects that require sound planning and significant oversight include new quality management system (QMS) and technical documentation requirements.
undertaking. As many changes need to be made in the quality system and business processes to meet the new regulations, some organizations are experiencing challenges in implementing and demonstrating compliance to their notified bodies. To start, organizations need to ensure that their devices have the accurate certificate scopes and that current certificates can be maintained until expiration. Several significant important aspects that require sound planning and significant oversight include new quality management system (QMS) and technical documentation requirements.
This is the first article in a two-part series. This article highlights the sections that need close attention regarding economic operator responsibilities, overall QMS considerations, and common challenges during EU MDR and EU IVDR implementation to ensure a smooth transition and avoid pitfalls during implementation. The second article will focus on typical challenges experienced during the technical documentation assessments conducted by the notified bodies.
Economic Operators’ Responsibilities Across The Supply Chain
Accountability across economic operators is significantly increased and summarized in the table below:
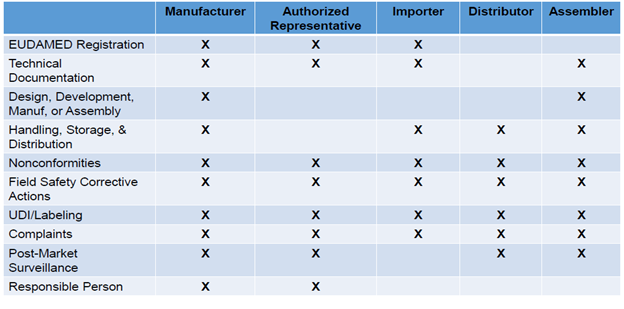
Information related to the economic operator (type), contact details, and basic Unique Device Identification-device identifier (UDI-DI) information (risk class, notified body information) must be well defined. EU MDR and EU IVDR require assigning a basic UDI-DI to the device and providing it to the UDI database together with other core elements. Additionally, manufacturers need to verify in the European database on medical devices (EUDAMED) that the information is accurate and up to date.
The new regulations require the European Commission to manage an electronic system to create a single registration number (SRN) that identifies manufacturers, authorized representatives, and importers. The Member States maintain the registration of distributors of devices that have been made available in their territory. Importers are required to verify that the manufacturer or authorized representative has provided the required information to the electronic system and notify the authorized representative or manufacturer of any discrepancies. SRNs are obtained from the competent authorities and are used to apply for conformity assessment with a notified body and to access EUDAMED. Data is accessible by the public and is used by the competent authority to determine required fees. Data must be updated regularly and verified for accuracy to comply with the regulation.
It is also important to keep in mind that initial audits must be done at least partially on-site. This has become challenging due to current COVID-19 restrictions; however, manufacturers need to begin contingency plans and formalize agreement with their notified bodies to address this situation. MDCG 2020-4 provides guidelines on addressing this time-limiting factor; read my article here for discussion on this topic.
QMS Requirements That Require Careful Review And Consideration
New documentation and record-keeping requirements: Many existing procedures need to be updated, starting with the quality manual, which must reference the new regulations, new common specifications, and standards. If an intended purpose needs to be adjusted, the Medical Device File and other documents will also need to be updated. New record retention requirements shall also be assessed for all devices.
According to Article 10 of both EU MDR and EU IVDR, manufacturers shall keep the technical documentation, the EU declaration of conformity, and, if applicable, a copy of the relevant certificate, including any amendments and supplements — issued in accordance with Article 56 for MDR and Article 51 for IVDR — available for the competent authorities for a period of at least 10 years after the last device covered by the EU declaration of conformity has been placed on the market.
Person responsible for regulatory compliance: Manufacturers and authorized representatives shall have available within their organization at least one person responsible for regulatory compliance who possesses the requisite expertise in the field of medical devices or in vitro diagnostic devices, as applicable. The specific expertise and qualification requirements can be found in Article 15 of EU MDR and Article 15 of EU IVDR.
Strategy for regulatory compliance: A strategy for regulatory compliance that includes how the organization will comply with conformity assessment procedures and how it will manage modifications to the devices must be in place. This strategy shall also include processes for identification of relevant legal requirements, qualification, classification, and handling of equivalence. Quality plans outlining the IVDR transition and the plans for updating the quality system or the technical documentation can demonstrate compliance with this requirement. Establishing continual improvement processes implemented according to Article 10 of EU MDR and EU IVDR should also be taken into consideration as part of the strategy for regulatory compliance.
Implant card: Requirements are outlined in Article 18 of EU MDR.
UDI: Requirements are outlined in Article 27 and Annex VI of EU MDR and Article 24 and Annex VI of EU IVDR.
New plans and strategies: The table below summarizes the plans required by MDR and IVDR that are subject to QMS audits and technical review assessments:
|
MDR |
IVDR |
|
Strategy for Regulatory Compliance – Article 10 & Annex IX |
Strategy for Regulatory Compliance – Article 10 & Annex IX |
|
Risk Management Plan – Annex I |
Risk Management Plan – Annex I |
|
Clinical Development Strategy – Article 61 |
|
|
Clinical Evaluation Plan – Annex II, Annex IX, Annex XIV, Annex XV |
Performance Evaluation Plan – Annex IX, Annex XIII |
|
Clinical Development Plan – Annex XIV |
Clinical Performance Study Plans – Annex XIII, Annex XIV |
|
Clinical Investigation Plan – Annex XV |
Clinical Performance Study Plans – Annex XIII, Annex XIV |
|
Post-Market Surveillance Plan – Annex III |
Post-Market Surveillance Plan – Annex III |
|
Post-Market Clinical Follow-Up Plan – Annex XIV |
Post-Market Performance Follow-Up Plan – Annex XIII |
Clinical evaluation applicable to EU MDR: Article 61 indicates that for all class III devices and class IIb devices intended to administer and/or remove a medicinal product, the manufacturer may, prior to clinical evaluation or investigation consult an expert panel about its clinical development strategy. The manufacturer shall give due consideration to the views of the expert panel. This consideration shall be documented in the clinical evaluation report.
Summary of safety and performance: For implantable devices, class III devices, and in-vitro diagnostic devices class C and class D, manufacturers are required to write up a summary of safety and clinical performance in a way that is clear to the intended user and the patient. Notified bodies are required to review and submit this information to EUDAMED to make it available to the public. There are several requirements to be included by manufacturers in this summary, such as: basic UDI-DI information, SRN, device description and purpose, reference to harmonized standards, a summary of clinical and/or performance evaluation, relevant information on post-market follow-up, suggested training for users and information on residual risks, undesirable effects, warnings, and precautions, among other aspects. For in vitro diagnostic devices, metrological traceability of assigned values is also to be included in this summary.
Typical Challenges Witnessed During Implementation
Organizations must keep in mind that at the time of a QMS assessment to the MDR or IVDR, the quality management systems must be established, documented, and operational (where practical) for all device groups according to EU MDR and EU IVDR Article 10: General obligations of manufacturers. There are many requirements that notified bodies must carefully review now that could end up in non-conformances if they are not properly implemented. For example:
A Strategy For Regulatory Compliance, Including Compliance With Conformity Assessment Procedures And Procedures For Management Of Modifications To The Devices Covered By The System
It is common for organizations to inadvertently define a strategy for regulatory compliance that does not cover all the product types subject to the regulation or all the conformity assessment routes. To avoid a non-conformance here, it is important to conduct a thorough assessment to confirm that the strategy encompasses all applicable products in the scope of certification.
Additionally, being able to demonstrate control and monitoring of economic operators within the entire supply chain can be a regulatory compliance challenge during an audit. This can be successfully accomplished through documented quality agreements that outline new responsibilities for distributors, importers, assemblers, and authorized representatives but it’s important that they are formalized prior to an assessment. Each economic operator shall be able to check compliance of all the others involved. For example, distributors must verify the CE mark and EU declaration of conformity, labeling, instructions for use (IFU), and UDI; importers should be able to verify designated authorized representative and maintain details on labeling/packaging, while authorized representatives must verify technical documentation and conformity assessment from the manufacturer and have access to the declaration of conformity and technical documentation.
Identification Of Applicable General Safety And Performance Requirements And Exploration Of Options To Address Those Requirements
Organizations must conduct an impact assessment of new safety and performance requirements across the entire quality system and not manage them as isolated requirements. For example, risk management documentation must include the impact of these requirements, including mitigation activities.
Resource Management, Including Selection And Control Of Suppliers And Subcontractors
Qualifications and experience for the person responsible for regulatory compliance must be defined, and records supporting these criteria shall be available. If notified bodies cannot verify this information or conclude that the qualifications/experience are inadequate, the company would not be able to comply with this requirement. In addition to having the documentation available, organizations need to be able to demonstrate that the person responsible for regulatory compliance is permanently and continuously available to support them; this can be managed through a documented agreement in the case of subcontractors, but it is important to not lose sight of this requirement.
Additionally, it is important to show allocation of resources for transition and post-transition requirements in post-market surveillance and to ensure that the legal manufacturer holds all the technical documentation for the devices. This can be demonstrated through quality management review minutes and other records demonstrating resource management is being considered at senior levels in the organization.
Risk Management As Set Out In In Section 3 Of Annex I
Risk management cannot be implemented as an isolated process. To successfully demonstrate that risk management is properly implemented, conclusions from risk management must link to clinical performance data. Claims in the IFU must link to the scientific validity and the analytical and clinical performance data, which should also be referenced in the performance evaluation report (PER). It is important that organizations properly address benefit-risk on all hazards and that there is a clear understanding of the concept of hazard vs. harm.
For IVDR: Performance Evaluation, In Accordance With Article 56 And Annex XIII, Including Post-Market Performance Follow-Up (PMPF)
A PMPF is used to confirm safety and validity of a device on the market. If it is confirmed during performance evaluation and risk management activities that the device is safe, PMPF studies are not required, unless issues are revealed from post-market surveillance activities. An important aspect of PMPF is to confirm a benefit-risk ratio for the intended purpose of the device has not been adversely affected.
A performance evaluation should be conducted through an objective review and must consider both favorable and unfavorable data. The depth and extent of the evaluation must be proportionate and appropriate to characteristics of the device, including risks, risk class, performance, and intended purpose. The outputs of the evaluation should lead to a plan for PMPF. If it is concluded that PMPF studies are not needed, this must be justified in the PER.
The performance evaluation shall be a continual process driven by a performance evaluation plan. The intended use is important and critical for setting the clinical evidence required, and scientific validity should link to the claims being made. This is required for all IVD devices, even if devices have been in the market for a long time. Articles 10 and 56 and Annex XIII of the IVDR address all the specific requirements.
Verification Of The UDI Assignments Made In Accordance With All Relevant Devices And Ensuring Consistency And Validity Of Information Provided
Organizations need to ensure that the UDI can be verified throughout the supply chain. Additionally, they need to verify that it is included in all the required locations, such as, for example, technical documentation, declaration of conformity, implant card, labeling/packaging, and vigilance reports. A UDI guidance document is available; however, it is mainly focused on medical devices. Therefore, it is important for in-vitro diagnostic manufacturers to assess whether self-tests, near patient tests, companion diagnostics, and class D devices need to implement UDI requirements.
Setting Up, Implementing, And Maintaining A Postmarket Surveillance System
Post-market surveillance shall be considered over the full product life cycle, such as design, manufacturing, shelf-life, lifetime, and disposal. Procedures should be implemented for post-market surveillance and post-market performance follow-up (for IVDR), with specific frequency requirements for each device class. Vigilance reporting requirements include implementation of systems for serious incidents, field safety corrective actions, and trend reports.
Organizations must have procedures in place to address reporting requirements, including sound statistical methodologies for monitoring vigilance trends. For example, serious public health threats must be reported within two days, death or unanticipated serious deterioration in the state of health must be reported within 10 days, and any other incidents within 15 days.
Periodic Safety Update Reports (PSURs) and Summary of Safety and Clinical Performance (SSCP) are mandatory reports that must be submitted at different frequencies as summarized below:
|
Device |
Periodic Safety Update Report (PSUR) |
Summary of Safety and Clinical Performance (SSCP) |
|
Class I |
PMS report updated when necessary |
|
|
Class IIa |
As necessary; at least every 2 years |
|
|
Class IIb |
Annual |
|
|
Class IIb Implantable |
Annual to Notified Body through EUDAMED |
Annual to Notified Body through EUDAMED |
|
Class III |
Annual to Notified Body through EUDAMED |
Annual to Notified Body through EUDAMED |
|
IVD A |
|
|
|
IVD B |
|
|
|
IVD C |
|
As soon as possible, where necessary |
|
IVD D |
|
As soon as possible, where necessary |
Organizations shall verify that procedures are updated to ensure these activities take place at the required frequencies and that agreements with all applicable economic operators are updated accordingly to avoid potential non-conformances.
Medical device organizations are now required to have several systematic processes that are subject to many new EU requirements, which focus mainly on safety and performance. Economic operators across the entire supply chain must apply faster product development cycles, maintain quality, and remain compliant with industry regulations. While there are many new specific requirements that could be inadvertently missed and a significant number of resources are needed to implement the new requirements, organizations can benefit from following guidelines and by adequately organizing data in their quality management system to provide a clear correlation of the regulation requirements and how the organization complies with them for each device. Additionally, preparing and organizing the technical documentation needed for EU MDR and EU IVDR compliance have their own unique challenges; the next part of this series will explore them, including some considerations to avoid pitfalls during the notified body assessments.
About The Author:
 Marcelo Trevino is the global vice president, regulatory affairs and quality assurance at Agendia, a molecular diagnostics company focused on breast cancer genomic testing. He has more than 25 years of experience in quality and regulatory affairs, serving in senior leadership roles with different organizations while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, and medical imaging/surgical navigation, amongst others. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR, EU IVDR, MDSAP). Trevino holds a B.S. in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. He is also a certified Quality Management Systems Lead Auditor by Exemplar Global. He can be reached at marcelotrevino@outlook.com or on LinkedIn.
Marcelo Trevino is the global vice president, regulatory affairs and quality assurance at Agendia, a molecular diagnostics company focused on breast cancer genomic testing. He has more than 25 years of experience in quality and regulatory affairs, serving in senior leadership roles with different organizations while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, and medical imaging/surgical navigation, amongst others. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR, EU IVDR, MDSAP). Trevino holds a B.S. in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. He is also a certified Quality Management Systems Lead Auditor by Exemplar Global. He can be reached at marcelotrevino@outlook.com or on LinkedIn.