
Improving Execution Of Biological Evaluation Of Drug Delivery Devices
By BioPhorum

The development of combination products, particularly drug–device integrations such as prefilled syringes, autoinjectors, and inhalers, has revolutionized therapeutic delivery by enhancing patient adherence, dosing precision, and self-administration. However, these innovations bring complex regulatory and biological safety challenges that demand harmonized risk-based approaches to ensure patient safety.
We have identified several challenges in the biological safety assessment of drug delivery devices, including the need for clarity and consensus in regulatory expectations, minimizing unnecessary testing, and improving supplier data quality.
This article outlines a selection of problems, impacts, goals, and benefits to address these challenges, aiming to streamline the process, reduce testing burdens, and improve patient safety:
- Problems — potential challenges and issues to be discussed and resolved, e.g., ambiguity and lack of clear guidance for combination drug delivery products in biological risk assessments
- Impacts — potential risks and negative effects these problems could have on organizations, e.g., problems lead to submissions that carry risk, due to uncertainty around applicable standards
- Goals — the objectives the team aims to achieve and deliver, e.g., create clear guidance on how to execute the biological safety assessment and optimize process flows for combination products
- Benefits — what the key companies involved in biological evaluations (such as biopharmaceutical organizations, device suppliers, and contract research organizations) will gain from collaboration on the proposed goals, e.g., reduced time-to-market.
Biological Risk Assessment
The initial step in establishing the biocompatibility profile of a device is to conduct a biological risk assessment (BRA) or biological evaluation. This refers to both the documentation and the process used to assess the product or material under consideration. This includes the biocompatibility requirements for the device or component based on the duration and type of contact with the patient, as well as any justifications for not conducting the recommended tests required to demonstrate the absence of additional biological hazards or impacts on patient safety. This risk-based process ensures that devices do not pose undue risks to patients and that they align with regulatory requirements and promote safety.
A biological safety risk assessment is a risk-based approach to understand potential risks, review current data, and identify any residual gaps. It determines if testing may be one of the next steps, if justified. The risk assessment encompasses the biological evaluation plan (BEP) and the biological evaluation report (BER). Figure 1 summarizes the biological evaluation process.
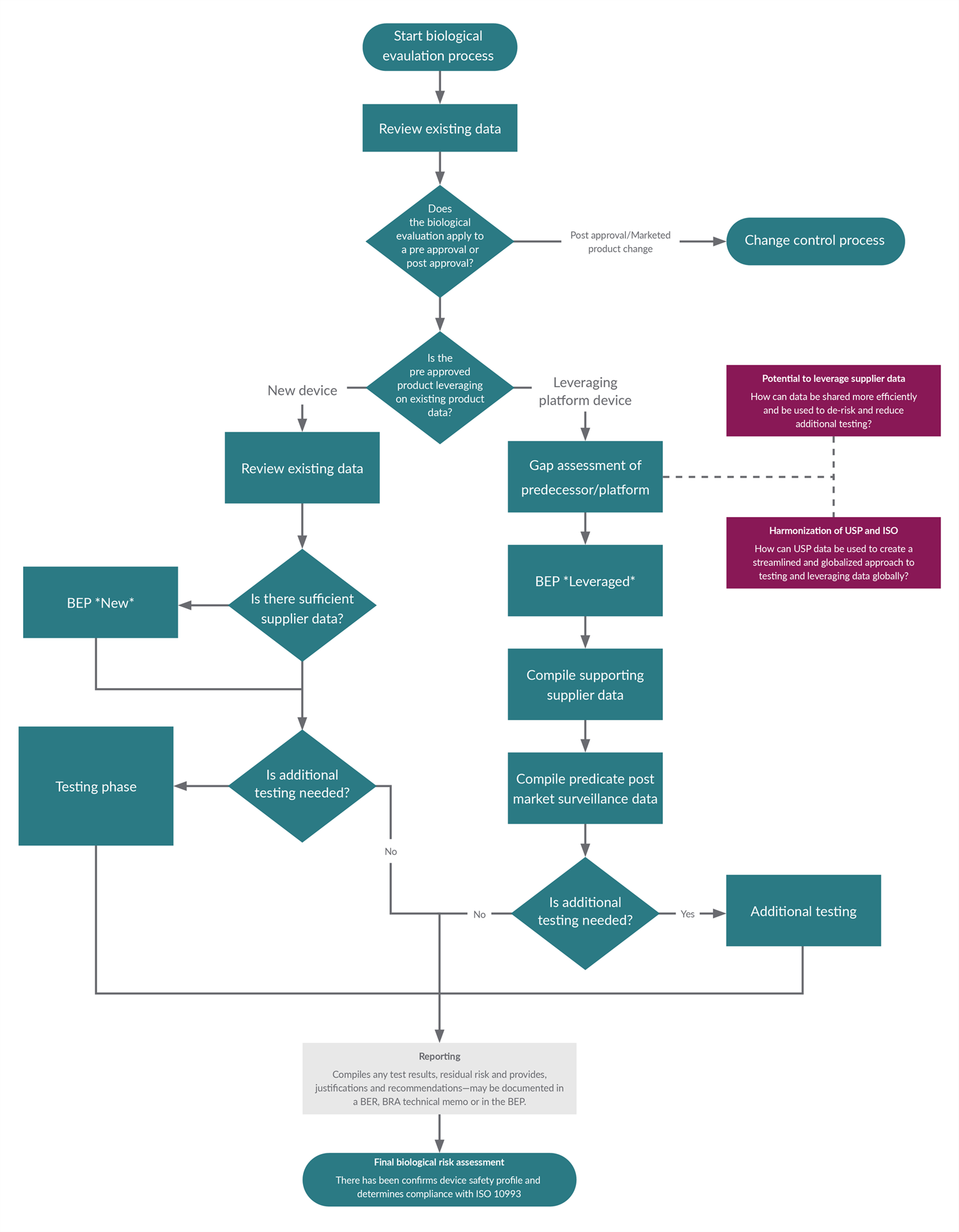
Figure 1: Biological evaluation process. Click on image to enlarge.
Problems include:
- Drug and device standards are established separately, which creates ambiguity for combination drug delivery products.
- While some harmonization exists (e.g., USP and ISO alignment), gaps remain, leaving limited direction for products that combine drugs and devices.
- Definitions of “medical device” differ across regions (such as the U.S. and EU), leading to inconsistent classifications (e.g., in certain circumstances, components could be considered container closure and/or medical device).
Impacts include:
- Lack of clear guidance for combination products leads to submissions that carry risk, due to uncertainty around applicable standards.
- Unnecessary animal testing raises ethical concerns, particularly when physical, chemical, in silico, and in vitro assessments could provide sufficient data.
- More resources are required by organizations.
Goals include:
- Streamline the process for creating a BEP.
- Create clear guidance on how to execute the biological safety assessment and optimize the process flow for combination products.
- Provide the industry with clarity on pitfalls when creating a platform approach for biological safety assessment.
Benefits include:
- reduced time-to-market
- more efficient use of resources
- improved patient safety.
Leveraging Supplier Data And Supplier Quality Interactions
Biological evaluation is a key design verification activity embedded in a broader risk management framework, as outlined in ISO 10993-1. A critical early step in this process is to gather physical and chemical information about the device or components.
Suppliers are often key contributors to biological evaluations by providing biocompatibility data and other safety-related documentation. This information can support material selections and facilitate the regulatory review process. In many cases, however, supplier-provided data is limited.
Problems include:
- Supplier-executed studies comply with older standards.
- Lack of industry consistency in supplier compliance statements regarding testing conformity.
- Supplier test data can be difficult to leverage due to reports lacking information on standard and detailed results of testing.
Impacts include:
- repeated testing due to a lack of information from suppliers
- delays in submission and subsequent approval
- misalignment of expectations can result in strained relationships between suppliers and license holders.
Goals include:
- Create a standardized supplier template based on data expectations, which can be used to assess the supplier.
- Create a best practice template for supplier selection.
- Set industry expectations around the data suppliers should provide.
Benefits include:
- minimized risk when going into submission
- reduced patient risk
- improved time-to-market.

ISO Versus USP: What Is The Expectation?
The link between USP and ISO testing requirements is unclear and inconsistent regarding combination products. There are instances when companies have successfully leveraged USP data to satisfy ISO requirements and others where this has failed. It appears to depend on the reviewer involved and the type of product. For biocompatibility, this would include standards such as USP <1663>, <1664>, <87>, <88>, and <1031>, as well as the ISO 10993 series.
Problems include:
- Lack of clarity on which guidance to follow, with significant differences between ISO and USP extractable testing methods, timeline, and costs.
- Reviewers often challenge the extractables testing methods of the CROs, including solvent selection, instrumentation, dilution, concentration methods, and recovery standards.
- Uncertainty regarding which components must comply with specific standards.
Impacts include:
- over-testing to meet the requirements of all standards without leveraging data
- delays in submissions and subsequent approvals
- inconsistent pushback from regulators when data has been leveraged with justification in place.
Goals include:
- clear understanding of the overlaps between USP and ISO 10993 standards during biological evaluations
- industry clarity around whether it is a component or a part of the device
- industry clarity on chemical characterization data.
Benefits include:
- reduced likelihood of pushback from regulators
- reduced resources
- reduced burden of testing.
Component–Device Categorization For Biological Safety Risk Assessment
Medical device categorization for biocompatibility risk assessments focuses on the nature and duration of body contact, which is critical for determining risk and identifying the required biological endpoint tests to establish the device’s biocompatibility. It is important to consider cumulative exposure when calculating the duration of body contact. However, ISO 10993-1 does not provide detailed guidelines for determining the duration of contact for repetitive single-use combination devices.
Problems include:
- no industry consensus/clear guidance on how to categorize combination products
- inconsistency in regulatory approval around the chosen category
- inconsistency in categorization recommendations from CROs.
Impacts include:
- over-testing due to the selection of a conservative category
- additional animal, environmental, and ethical concerns around over-testing
- additional time and cost associated with over-testing.
Goals include:
- industry consensus/clarity around combination product categorization based on requirements for the EU and the U.S.
- industry consensus/clarity around best practices for conducting cumulative calculations
- clarity around cumulative calculation for combination delivery devices.
Benefits include:
- reduced uncertainty in device development and program risk
- reduced approval times
- reduced testing leading to increased animal welfare and fewer ethical concerns.
Conclusion
The development and regulatory approval of combination products, particularly drug-device combinations, present unique challenges and opportunities for the biopharmaceutical industry. While the risk of a delivery device is generally low, ensuring biological safety through rigorous assessment and adherence to international standards, such as ISO 10993, is crucial to patient safety.
By streamlining processes, leveraging supplier data, and clarifying regulatory expectations, the industry can reduce testing burdens, accelerate time-to-market, and ultimately improve patient outcomes.
To achieve these goals, it is imperative for stakeholders across the biopharmaceutical industry to actively participate in collaborative initiatives. Companies should engage in open dialogue, share real-world challenges, and contribute to developing consensus views and best practices. By working together, we can create a more efficient and effective regulatory environment, minimize unnecessary testing, and enhance the safety of combination products.
This article summarizes the main points from a recent BioPhorum publication on this topic. To learn more, check out the full paper, Biological evaluation of drug delivery devices: unraveling the paradox.