
Introduction To ASTM E3263-20: Standard Practice For Qualification Of Visual Inspection Of Pharmaceutical Manufacturing Equipment And Medical Devices For Residues
By Andrew Walsh, Ralph Basile, Ovais Mohammad, Stéphane Cousin, Mariann Neverovitch, and Osamu Shirokizawa
Part of the Cleaning Validation for the 21st Century series
Visual inspection has been widely used for many years by the pharmaceutical, biologics, and medical device industries after cleaning to release manufacturing equipment and devices. However, visual inspection has never been demonstrated to be an effective, reliable, or safe method to use for these inspections. Recently, the European Medicines Agency (EMA) issued a Q&A1 to its guideline on determining health-based exposure limits (HBELs) that describes what criteria have to be met for visual inspection to be acceptable to the EMA for release of manufacturing equipment. Some form of guidance or a standard has been needed to guide these industries on how to meet these criteria and demonstrate that operators/QA inspectors are capable and qualified to accurately assess the absence or presence of residues on manufacturing equipment or medical devices. This article discusses the development and publication of a new ASTM International (American Society for Testing and Materials) standard practice for the qualification of visual inspection.
History And Regulatory Perspectives On Visual Inspection
U.S. regulation has required the “inspection of manufacturing equipment immediately before use” since 1979.2 While this regulation did not specify that the inspection should be "visual," in practice, pharmaceutical manufacturers have been releasing equipment based on a visual inspection for many years and industry and regulators have come to see this inspection as a visual inspection requirement. However, throughout all this time only a few studies on visual inspection have been performed, with varying results reported. Another misinterpretation of this statement has resulted in many companies only inspecting the equipment "immediately before use," after the cleaned equipment has been reassembled. After reassembly, large portions of equipment surfaces may not be visible, so they cannot be inspected and confirmed as visually clean. Visual inspection should take place after the cleaning process and before any reassembly of equipment.
In 1993, an article3 was published that mentioned that spiking studies indicated that many compounds were visible at approximately "100 μg per 2 x 2-inch swab area" (or approximately 4 μg/cm2). This 4 μg/cm2 value was quickly adopted by many companies as an "industry standard," although no data or any other supporting information were provided by the authors. Another article following in 1994 claimed that residues could be seen down to 1 μg/cm2 by using an additional light source.4 A subsequent article in 2000 claimed to see residues down to approximately 0.4 μg/cm2 for several compounds.5 A series of studies6,7 examining several different compounds found a range from 0.4 to >10 μg/cm2. Three studies using a different spiking technique that spread the residue evenly over the surface found detection limits for one residue at levels of 3, 5, and 7 μg/cm2. These detection limits were calculated following an ICH Q28 approach and were found to be influenced by several factors, including training.9 In 2010, Ovais Mohammad proposed using a statistical approach (logistic regression) to these spiking studies to more accurately derive the visible threshold.10
While extensive studies and analysis of the ability of visual inspection to identify the presence of residues have not been performed to date, regulatory agencies appear to be more flexible with regard to its use. The EMA's 2015 update to Annex 1511 now states that "It is not generally acceptable for this criterion alone to be used," indicating that visual inspection could be used alone for cleaning validation under certain circumstances. Aware of the significance of this statement, the recent ASTM E3106 Standard Guide12 provided the following guidance to support this Annex 15 statement:
"Using visual inspection alone for validation may be acceptable only when a Risk Assessment has shown that the risk is low and 100 percent of the equipment surface can be inspected under appropriate viewing conditions." (emphasis added)
However, it should be understood that regulators are highly unlikely to accept visual inspection alone for cleaning validation unless manufacturers have exceptional justification, such as a very low-hazard product, and will still likely expect some analytical testing to confirm acceptable cleaning during the cleaning validation phase. Where visual inspection can most likely be used alone is in subsequent cleaning process validations (verifications) for new low-risk products on multi-use equipment where prior satisfactory validation studies of the cleaning process have already been performed.
Moving even further in this direction, Pharmaceutical Inspection Co-operation Scheme (PIC/S)13 has now stated that “...spiking studies should determine the concentration at which most active ingredients are visible,” indicating that these health agencies are expecting to see visual inspection being used more frequently as a semi-quantitative tool and have requirements for its use. This statement specifically about active ingredients (APIs) has led some manufacturers to set up visual inspection and the related training activities to focus only on active ingredients. Any compound that is identified as a hazard in the Risk (Hazard) Identification step and found to be a risk in the Risk Analysis step may need to be included in a visual inspection qualification and training program. This is especially true for medical device manufacturing, as cleaning agents and processing aids may be the compounds identified as risks and not APIs.
Expanding further on this, on April 16, 2018 the EMA posted an update to its draft Q&A on the guideline for setting health-based exposure limits. In this final version, two new questions and answers appeared (Q7 and Q8) that are directly applicable to the use of visual inspection. These Q&As state:
Q7. Is analytical testing required at product changeover, on equipment in shared facilities, following completion of cleaning validation?
A: Analytical testing is expected at each changeover unless justified otherwise via a robust, documented Quality Risk Management (QRM) process. The QRM process should consider, at a minimum, each of the following:
• the repeatability of the cleaning process (manual cleaning is generally less repeatable than automated cleaning);
• the hazard posed by the product;
• whether visual inspection can be relied upon to determine the cleanliness of the equipment at the residue limit justified by the HBEL.
Q8. What are the requirements for conducting visual inspection as per Q&A 7?
A. When applying visual inspection to determine cleanliness of equipment, manufacturers should establish the threshold at which the product is readily visible as a residue. This should also take into account the ability to visually inspect the equipment, for example, under the lighting conditions and distances observed in the field.
Visual inspection should include all product contact surfaces where contamination may be held, including those that require dismantling of equipment to gain access for inspection and/or by use of tools (for example mirror, light source, borescope) to access areas not otherwise visible. Non-product contact surfaces that may retain product that could be dislodged or transferred into future batches should be included in the visual inspection.
Written instructions specifying all areas requiring visual inspection should be in place and records should clearly confirm that all inspections are completed.
Operators performing visual inspection require specific training in the process including periodic eye sight testing. Their competency should be proven through a practical assessment.
So, the regulatory requirements for implementing visual inspection as one of the tools available for cleaning validation are now pretty well defined by the EMA and all that was needed was detailed guidance on how to satisfy these criteria. The new ASTM 3263 was written specifically to provide the necessary guidance for establishing qualified visual inspection programs to comply with these newly clarified regulatory expectations.
ASTM E55 And F04 Committee Collaboration
In 2017 members of the ASTM E55 Committee on Manufacture of Pharmaceutical and Biopharmaceutical Products and members of the ASTM F04 Committee on Medical and Surgical Materials and Devices collaborated on writing a new Standard Guide that resulted in the E3219 Standard Guide for Derivation of Health Based Exposure Limits (HBELs).14 The team that developed the E3219 also included members of the E310612 team. The collaboration on the E3219 had been successful, and this team discussed collaborating on other standards that would benefit both pharmaceutical and medical devices and other industries and, in particular, for the qualification of visual inspection.
Therefore, in March of 2019, a new Work Item (WK67425) was initiated on the ASTM website and a collaboration area for the WK67425 was created. The original E3219 and E3106 teams were expanded to include several more pharmaceutical and medical device industry stakeholders to work on this standard (Table 1).
Table 1: WK67425 Collaboration Area Team Members
|
Team Member |
Company |
Industry Expertise |
|
Ralph Basile |
Healthmark Industries Co |
Medical Device |
|
Dhanapal Boopathy |
Zimmer Biomet |
Medical Device |
|
Stéphane Cousin |
GSK Vaccines |
Pharmaceutical |
|
Delane Dale |
Confluent Medical Technologies |
Medical Device |
|
Parth Desai |
Nostrum Laboratories Inc. |
Pharmaceutical |
|
Jayen Diyora |
Alnylam Pharmaceuticals |
Pharmaceutical |
|
Christophe Gamblin |
Theraxel |
Pharmaceutical |
|
Igor Gorsky |
ConcordiaValsource, LLC |
Pharmaceutical |
|
Jove Graham |
Geisinger Center for Health Research |
Medical Device |
|
Jessica Graham, Ph.D., DABT |
Bristol Myers Squibb |
Pharmaceutical |
|
Barbara Kanegsberg |
BFK Solutions LLC |
Medical Device |
|
Reto Luginbuehl |
Blaser Swisslube AG |
Medical Device |
|
Spiro Megremis |
American Dental Association |
Medical Device |
|
Mariann Neverovitch |
Bristol-Myers Squibb |
Pharmaceutical |
|
Mohammad Ovais |
Pharmaceutical Consultant |
Pharmaceutical |
|
Rodney Parker |
Stryker |
Medical Device |
|
Vimal Sachdeva |
World Health Organization |
Pharmaceutical |
|
Stephen Spiegelberg, Ph.D. |
Cambridge Polymer Group |
Medical Device |
|
Norma Turner |
Cambridge Polymer Group |
Medical Device |
|
Andrew Walsh |
Center for Pharmaceutical Cleaning Innovation |
Pharmaceutical |
Goals Of ASTM E3263
The goals for this new standard practice are to specifically provide instructions for qualification of visual inspection for residues on pharmaceutical manufacturing equipment and medical devices. This new standard would provide guidance for achieving the following six goals:
- An approach that applies the science-based, risk-based, and statistics-based concepts and principles introduced in Guides E310612 and E3219.14
- An approach for qualifying the inspection of equipment for cleanliness in accordance with 21 CFR 211.67(b).2
- An approach for qualifying the visual inspection of equipment for cleanliness in accordance with European Medicines Agency (EMA) Annex 15.11
- An approach for qualifying the visual inspection (and visual threshold) of equipment for cleanliness in accordance with the EMA’s Q&A Guidance (Q&A 7 and Q&A 8).1
- An approach that would apply the risk-based principles introduced in ICH Q915 so that the level of effort, formality, and documentation for cleaning validation would also be commensurate with the level of risk.
- An approach for releasing manufacturing equipment and manufactured medical devices or cleanliness that is compatible with the U.S. FDA’s guidance on its Process Analytical Technology Initiative.16
Scope Of ASTM E3263
The E3263 provides statistically valid procedures for determining the visual detection limit (also called the visual threshold) of residues and for the qualification of operators/QA inspectors to perform the visual inspection of pharmaceutical manufacturing equipment surfaces and medical devices. E3263 applies to pharmaceuticals, including active pharmaceutical ingredients, finished dosage forms, over-the-counter (OTC) drugs, veterinary medicines, biologics, clinical supplies, medical devices, cosmetics, and consumer products. E3263 can be used for all types of chemical residues (including APIs, biological substances, intermediates, cleaning agents, processing aids, machining oils, etc.) that could remain on manufacturing equipment surfaces or the surfaces of medical devices.
The E3263 Standard Practice Guide
The Procedure section of E3263 contains guidance on six main elements. The first element discusses what initial criteria must be met in order to implement a visual inspection program.
1. Initial Criteria for Establishing Qualification Programs for Visual Inspection:
- Calculation of MSSR – MSSRs (Maximum Safe Surface Residues) must first be calculated for all equipment to be inspected, as it is necessary to determine the minimum level of residue that must be detectable by the visual inspection. The MSSR, expressed in mass units per surface area (for example, μg/cm2), is calculated using the following equation (from ASTM E3106):

- Selection of Surfaces for the Qualification Study – the steps to take in selecting the materials of construction for visual inspection studies
- Selection of Products/Compounds for the Qualification Study – the steps to take in selecting the compounds/products (e.g., APIs, cleaning agents, machining oils, etc.) for visual inspection studies
- Preparation of Surrogate Surfaces or Devices – how to prepare the surrogate surfaces (e.g., coupons, equipment parts, medical devices, etc.) for use in visual inspection studies
- Surrogate Surface Storage and Handling – how the surrogates’ surfaces should be handled
- Viewing (Lighting) Conditions – what the lighting requirements are for performing the visual inspection studies
2. Inspector Training – This section discusses what is necessary to train operators/QA inspectors for visual inspection and maintain their qualified state.
3. Determination of Visual Residue Limits (VRL) – This section discusses how to identify the lowest spiked residue level (visual threshold) that is most likely to be seen by all trained operators/QA inspectors for the product/compound of a spiked coupon study. This spiked residue level (visual threshold) should be the starting point for inspector qualification studies. In cases where the VRL is determined in a study using a small number of inspectors (e.g., N=4) the VRL may not be statistically justifiable. A method for setting scientifically and statistically justifiable VRLs using logistic regression analysis (Figure 1) was developed by Ovais Mohammad to provide a meaningful determination of the VRL.17
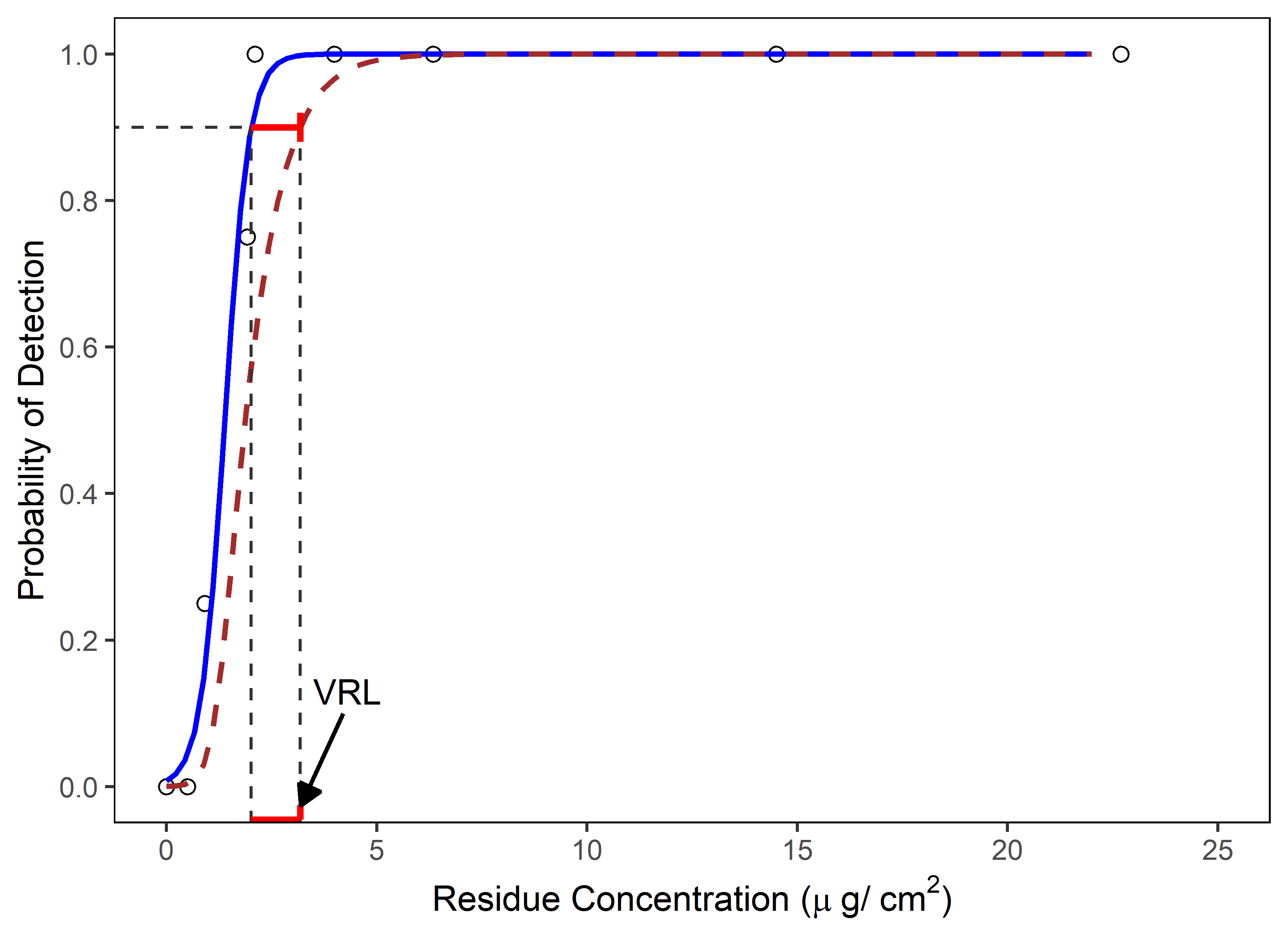
Figure 1 – Determination of VRL using Logistic Regression Analysis: The solid blue line is predicted probability of detection and the brown dashed line is the lower 95 percent confidence bounds for the predicted probabilities. In this example, VRL represents the residue concentration at the lower 95 percent confidence for 90 percent probability of detection. (Reprinted from ASTM E3263-20 "Standard Practice for Qualification Of Visual Inspection Of Pharmaceutical Manufacturing Equipment And Medical Devices For Residues", copyright ASTM International, 100 Barr Harbor Drive, West Conshohocken, PA 19428, USA, www.astm.org.)
4. Qualification of Operators/QA inspectors Using Attribute Agreement Analysis – This section discusses how to set up a visual inspection study to qualify any number of operators/QA inspectors by analyzing inspection results as attribute/binary data (i.e., clean/dirty).
5. Acceptance of the VRL for Cleaning Validation – This section discusses how to determine whether visual inspection is appropriate for use by comparison to the MSSR using the Visual Detection Index (VDI).18
6. Documentation – This section discusses what key documents are necessary for establishing a visual inspection program.
Significance Of HBELs For Visual Inspection
Probably the most important consideration for the implementation of visual inspection is the HBEL of the compound being considered. The 1/1,000th of a dose and 10 ppm limits have been shown to be overly conservative with low-hazard compounds and not restrictive enough with high-hazard compounds, which does not reflect the risk-based approach of ICH Q9.18 This error in logic extends to the use of visual inspection. A simple analysis will demonstrate this.
The MSSRs for the 304 drug compounds in the article cited above19 were calculated for both HBELs and the 1/1,000th of a dose / 10 ppm combination using the parameters shown in Table 2. The total equipment surface area was chosen to be typical of a packaging line, which can be considered one of the more appropriate areas for visual inspection.
Table 2: Parameter Assumptions for MSSR Calculations
|
Parameter |
Value |
|
Batch Size |
100 kg |
|
Maximum Daily Dose |
10 g |
|
Total Equipment Surface Area |
25,000 cm2 |
The MSSRs for both HBELs and the 1/1,000th of a dose / 10 ppm combination were plotted as shown in Figure 2. A reference line for a visual residue limit at 10 µg/cm2 has been added and a box drawn to contain the compounds that are below or possibly too close to this visual residue limit to allow for visual inspection. It should be obvious from this graph that many, if not most, compounds could not be considered for visual inspection.

Figure 2 – Comparison of MSSRs for HBELs and 1/1,000th & 10 ppm: The о symbols represent the MSSRs based on HBELs and the ∆ symbols are their corresponding MSSRs calculated from the 1/1,000th or 10 ppm limits. (DL = Detection Limit) X-axis indicates the number of compounds. Data plotted in the "R" statistical programming language by Ovais Mohammad.
Figure 3 shows the same graph with the ∆ symbols for the MSSRs for the 1/1,000th of a dose / 10 ppm combination removed. The box is now drawn to contain the compounds that are above this visual residue limit and would allow for visual inspection. It should be obvious from this graph that, with the HBELs, there are many compounds, in particular low-risk compounds, that could possibly be considered for visual inspection.

Figure 3 – MSSRs for HBELs with 1/1,000th & 10 ppm MSSRs Removed: Data plotted in the "R" statistical programming language by Ovais Mohammad.
Summary
While the EMA's new Q&A 7 and Q&A 8 may have been a surprise for many in the industry, they were added to allow companies with products found to be low risk (based on their HBELs) the option of using visual inspection at product changeovers. Hopefully, from the discussion above, it is clear that companies must finally let go of holding on to the historical 1/1,000th dose and 10 ppm limits and implement the HBEL in order to take advantage of visual inspection.
At the same time, some companies have already started moving to visual inspection for release of equipment by simply stopping swab/rinse testing without any QRM program in place, any adequate justification, or any qualification of their operators/QA inspectors. These are unacceptable practices that will inevitably lead to regulatory action resulting in significant costs and reputational damage for these companies. Implementing procedures as described in E3263 should prevent this. However, E3263 cannot be implemented completely independent of the ASTM E3106 and E3219 standards and must be coordinated with these guides.
Again, as stated in the "History And Regulatory Perspectives On Visual Inspection" section, visual inspection alone will most likely find initial use in cleaning process validations on multi-use equipment for new low-risk products. At least initially, this will require that prior satisfactory validation studies of the cleaning process already exist. The 6-step QRM process described below would be an appropriate procedure to implement visual inspection.
- The HBEL of the new product must be derived by a qualified toxicologist and compared to the HBELs of the existing portfolio of products using the Toxicity Scale.20 If the hazard (toxicity) level is acceptable, the product can move to step 2.
- HBEL-derived cleaning (swab) limits should be calculated for the new product, compared to the existing cleaning data for the equipment, and its potential Process Capability Score (Cpu Score) calculated.21 If the Cpu Score is acceptable then the product can move to step 3.
- The "Cleanability" of the new product is measured and compared to the existing "Hardest-to-Clean" product.22 If the Cleanability of the new product is acceptable then the product can move to step 4.
- The visual detection limit (visual threshold) should be determined for the new product and the Visual Detection Index (VDI) calculated.18 If the VDI is acceptable then visual inspection alone could be justified.
- Qualification of all operators/QA inspectors to visually inspect the new product23
- Visual inspection of 100 percent of (disassembled) equipment surfaces should be performed and documented after each batch of the new product is manufactured.
The authors believe that the new E3263 standard provides the science-, risk-, and statistical-based guidance and the tools needed for companies to implement the use of visual inspection within a QRM program that meets the criteria promulgated in the EMA's new Q&A 7 and Q&A 8.
Peer Review
The authors wish to thank Thomas Altman, Joel Bercu, Ph.D., Sarra Boujelben, Alfredo Canhoto, Ph.D., Gabriela Cruz, Ph.D., Mallory DeGennaro, Parth Desai, Andreas Flueckiger, M.D., Christophe Gamblin, Ioanna-Maria Gerostathes, Ioana Gheorghiev, M.D., Jessica Graham, Ph.D., DABT, Crystal Hamelburg, Ester Lovsin-Barle, Ph.D., Miquel Romero Obon, Prakash Patel, Siegfried Schmitt Ph.D., and Joel Young for reviewing this article and for providing insightful comments and helpful suggestions.
References:
- European Medicines Agency, “Questions and answers on implementation of risk-based prevention of cross-contamination in production” and “Guideline on setting health-based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities,” 19 April 2018, EMA/CHMP/CVMP/SWP/246844/2018.
- 21 CFR 211.67 Equipment Cleaning and Maintenance
- Fourman, G. L., and Mullen, M. V., “Determining Cleaning Validation Acceptance Limits for Pharmaceutical Manufacturing Operations,” Pharmaceutical Technology, 17 (4) 1993
- Jenkins, K. M. and Vander Weilen, A. J., “Cleaning Validation: An Overall Perspective,” Pharmaceutical Technology, 18, (4) 1994
- Buscalferri, F., Lorenzen, S., Schmidt, M., Schwarm, H.-M., Anhalt, E., et al., “Bestimmung der Sichtbarkeitsgrenzen von pharmazeutischen Feststoffen auf Edelstahloberflächen,” Pharm. Ind., Vol 62, No. 6, 2000.
- Forsyth, R. J., Van Nostrand, V., and Martin, G. P., “Visible-Residue Limit for Cleaning Validation and its Potential Application in Pharmaceutical Research Facility,” Pharmaceutical Technology, 28 (10) 2004.
- Forsyth, R. J., and Van Nostrand, V., “Using Visible Residue Limits for Introducing New Compounds into a Pharmaceutical Research Facility,” Pharmaceutical Technology, 29 (10) 2005.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology - ICH Q2 (R1), www.ich.org.
- Desai, P., and Walsh, A., “Validation of Visual Inspection as an Analytical Method for Cleaning Validation,” Pharmaceutical Online, August 2017.
- Ovais, M., “Statistically Justifiable Visible Residue Limits,” Pharmaceutical Technology, 34 (3) 2010.
- EudraLex Volume 4 Guidelines for Good Manufacturing Practices for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation
- American Society for Testing and Materials E3106-18e1 "Standard Guide for Science Based and Risk Based Cleaning Process Development and Validation" www.astm.org.
- Pharmaceutical Inspection Co-operation Scheme (PIC/s), https:// picscheme.org.
- American Society for Testing and Materials E3219-20 "Standard Guide for Derivation of Health Based Exposure Limits (HBELs)" www.astm.org.
- International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, ICH Harmonized Tripartite Guideline, Quality Risk Management – Q9, Step 4, 9 November 2005, www.ich.org.
- FDA Guidance for Industry: PAT – A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance, September 2004, U.S. Food and Drug Administration (FDA), www.fda.gov.
- Ovais, Mohammad, “Statistically Justifiable Visible Residue Limits,” Pharmaceutical Technology, Vol 34, No. 3, March 2010.
- Walsh, Andrew, Thomas Altmann, Alfredo Canhoto, Ester Lovsin Barle, David G. Dolan, Mariann Neverovitch, Mohammad Ovais, Osamu Shirokizawa and Kelly Waldron. "An MSSR-derived Scale for Assessing the Detectability of Compound-Carryover in Shared Facilities" Pharmaceutical Online December 2017
- Walsh, Andrew, Michel Crevoisier, Ester Lovsin Barle, Andreas Flueckiger, David G. Dolan, Mohammad Ovais "Cleaning Limits—Why the 10-ppm and 0.001-Dose Criteria Should be Abandoned, Part II" Pharmaceutical Technology 40 (8) 2016
- Walsh, Andrew, Ester Lovsin Barle, Michel Crevoisier, David G. Dolan, Andreas Flueckiger, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "An ADE-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online May 2017
- Walsh, Andrew, Ester Lovsin Barle, David G. Dolan, Andreas Flueckiger, Igor Gorsky, Robert Kowal, Mohammad Ovais, Osamu Shirokizawa, and Kelly Waldron. "A Process Capability-Derived Scale For Assessing Product Cross-Contamination Risk In Shared Facilities" Pharmaceutical Online August 2017
- Song, Ruijin, Alfredo Canhoto, Ph.D., and Andrew Walsh "Cleaning Process Development: Cleanability Testing and "Hardest-To-Clean" Pharmaceutical Products" Pharmaceutical Online January 2019.
- American Society for Testing and Materials E3263-20 "Standard Practice for Qualification of Visual Inspection of Pharmaceutical Manufacturing Equipment and Medical Devices for Residues" www.astm.org.