
ISO TR 24971:2020 — Bringing Clarity To Risk Acceptability In ISO 14971
By Edwin Bills, Consultant
Periodically, standards are revisited by international and national committees to determine if they are still current or need revision or withdrawal. Through the voting process and based on comments received during the voting period in early 2016 on both ISO 14971:2007 (the standard for application of risk management to medical devices) and ISO TR 24971:2013 (the guidance on the application of ISO 14971), ISO determined that these documents needed to be updated and the policy for risk acceptability should be revised for clarity.
revision or withdrawal. Through the voting process and based on comments received during the voting period in early 2016 on both ISO 14971:2007 (the standard for application of risk management to medical devices) and ISO TR 24971:2013 (the guidance on the application of ISO 14971), ISO determined that these documents needed to be updated and the policy for risk acceptability should be revised for clarity.
Among the issues that needed attention was the requirement for top management responsibility for establishing a policy for determining risk acceptability criteria in clause 3.2 of ISO 14971:2007, which was addressed in the ISO 14971:2019 Third Edition. The policy for risk acceptability criteria has been a part of ISO 14971 since its inception in 2000, but many have raised questions about what this requirement means. Some have created risk matrices or risk charts to describe the requirement, but that is not a correct interpretation of the standard, and should not be used.
The resulting revision of ISO TR 24971:2013 — ISO TR 24971:2020 — provides extensive guidance in the informative annexes, discussions of the requirements in ISO 14971:2019, and further discussion of the terms “benefit” and “benefit-risk analysis.” It does not add any requirements. It is only guidance or help for those implementing the standard.
The Requirement
ISO 14971:2019 clause 4.2 requires that:
Top management shall define and document a policy for establishing criteria for risk acceptability. This policy shall provide a framework that ensures that criteria for risk acceptability are based on applicable national/regional regulations, relevant international standards, take into account the generally acknowledged state of the art, and known stakeholder concerns.
An additional note, which is informative and is not a requirement, provides more information regarding the approach to risk control in the risk acceptability policy:
NOTE 1 The manufacturer’s policy for establishing criteria for risk acceptability can define the approaches to risk control: reducing risk as low as reasonably practicable, reducing risk as low as reasonably achievable, or reducing risk as far as possible without adversely affecting the benefit-risk ratio. See ISO/TR 24971[9] for guidance on defining such policy.
These are the same requirements as in ISO 14971:2007 clause 3.2, and also clause 3.3(a) in the earlier 2000 edition. Unfortunately, these requirements were not further explained until ISO TR 24971:2013 was released. Even then, more information with examples might have helped in understanding this requirement, and that is what ISO TR 24971:2020 Annex C provides.
Past Incorrect Implementations
Because of a lack of direction from the technical committee in the earlier editions of the standard and the technical report, many interpretations of the requirement came into being. Among them was interpretation defining the acceptable risk as a risk matrix using a two-dimensional chart with one axis being identified as probability of occurrence of harm and the other axis being severity of harm with appropriate levels being chosen by the manufacturer and identified by notations along with the risk chart. Many could not agree on a standard appearance of such a matrix and it seemed every interpretation was different with the zero points being any of the four corners, and exchanging the severity and probability axes, though most often the zero point was the lower left or upper left corner. A line through the matrix was identified by the company as identifying the boundary between acceptability and unacceptability. This was most often, though not always, shown as the boundary between the Intolerable Risks and the Investigate for Further Risk Reduction regions, though other terms were used to identify the regions.
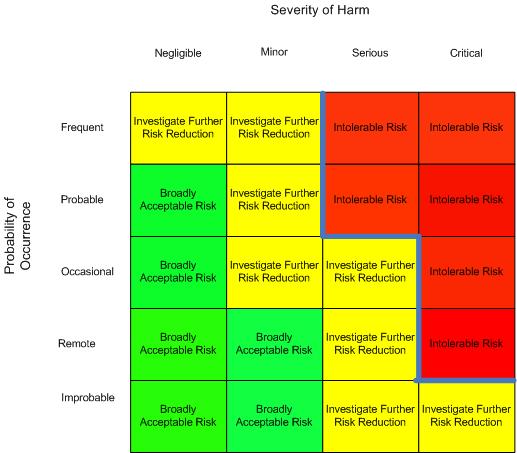
Figure 1: Risk Matrix with Acceptability
Some incorrectly used a technique to establish risk acceptability from earlier editions of failure modes and effects analysis (FMEA) identified as risk priority number (RPN). It is important to note the RPN technique was removed from the automotive industry FMEA as it was very confusing and inaccurate, and led to incorrect choices. It should not be used by the medical device industry to establish risk acceptability for much the same reasons. It is not establishing risk acceptability according to any of the risk acceptability approaches recognized in medical device risk management.
Instead of having Acceptable Risk and Unacceptable Risk regions defined by policy, some have added an intermediate region erroneously identified as As Low As Reasonably Practicable (ALARP). ALARP is not a region on a chart but is an approach for identifying the process of how far to reduce risk. Many companies continued to use this inaccurate interpretation at least until the release of the ISO 14971:2019 standard. The middle region is more correctly an Investigate Further Risk Control region, (ISO TR 24971:2020 Figure C1) meaning that risks that fall on a risk chart in this region should be further reduced by applying additional risk control measures.
In the European Union (EU), the confusion on acceptable versus unacceptable risks was muddied in the release of the EN ISO 14971:2012 edition, which indicated a company could not use the ALARP approach but should reduce risk using the As Far As Possible (AFAP) approach following the three medical device directives in Europe. The EN 2012 standard did not identify a process for identifying how the level required could be accomplished, causing more confusion. Providing objective evidence to auditors and regulators that AFAP has been reached is difficult, if not impossible. One more risk control could always be applied with some degree of improvement, even if it is infinitesimal. Making the decision of how much improvement is enough is difficult. The EN ISO 14971:2012 version was withdrawn by CEN with the release of EN ISO 14971:2019.
New Regulations
The recent Medical Device Regulations (MDR) and In Vitro Diagnostics Regulations (IVDR) regulations replacing the directives in the EU has not improved the situation to any great degree, requiring the manufacturer to reduce risks AFAP without impacting the benefit-to-risk ratio, yet the two regulations do not identify what a benefit-to-risk ratio is or how to accomplish reaching the AFAP goal. The term “benefit” is defined in ISO 14971:2019 3.2, but nowhere else in regulations, guidance, or standards. An extensive discussion, with examples, of “benefit” and “benefit-risk analysis” is found in ISO TR 24971:2020 7.4. This approach seems to indicate that risk charts may not be useful, as each risk must be reduced to AFAP on its own without consideration of acceptable risks.
ISO TR 24971:2020 Clarifications
A clarification in Annex C of ISO TR 24971:2020 indicates that individual risks may have different levels of risk acceptability than the overall residual risk. If a device has these different levels for the two types of risk — individual and overall residual — then these differing levels must be identified in the product risk management plan.
In addition, Annex C identifies five possible elements of the policy for risk acceptability criteria then provides a possible example for each of these elements:
- Purpose
- Scope
- Factors and considerations for determining risk acceptability criteria
- Approaches to risk control (e.g., ALARP, AFAP, As Low As Reasonably Achievable [ALARA], As Low As Possible [ALAP])
- Requirements for review and approval
The section requiring the most work to develop in the criteria for risk acceptability is the section on factors and considerations. It will require some effort to identify the appropriate elements for this section. There is guidance in Annex C to assist in this element.
“Approaches” is a decision point for the company management in selecting the appropriate approach. This decision may be influenced by the regulatory requirements, such as the EU’s use of AFAP (without impacting the benefit-to-risk ratio) in the MDR and IVDR.
To complete the risk acceptability criteria process, ISO TR 24971:2020 includes an additional set of examples comparing the elements of a policy, the acceptability criteria, and the evaluation of the results for the four elements in a policy:
- Regulatory requirements for the intended markets can be found and applied to the device risk acceptability criteria, but if the markets change, it is important to update the risk acceptability criteria with new market requirements.
- International standards that impact a product from the product-specific to the cross-cutting horizontal product safety standards must be considered in the development of the risk acceptability criteria; this might include electrical safety standards such as IEC 60601-1 and its family of standards.
- State of the art, which is a concept easily confused.*
- Stakeholder concerns can be collected from focus groups and product experts in the use of their particular product type in the environment of the intended use for their product type. It is important here to use the input from experts in the current use of the product based on current medical practice.
* ISO 14971:2019 attempted to end the confusion surrounding the term “state of the art” by providing a definition for the term.
3.28
state of the art
developed stage of technical capability at a given time as regards products, processes (3.14) and services, based on the relevant consolidated findings of science, technology and experience
Note 1 to entry: The state of the art embodies what is currently and generally accepted as good practice in technology and medicine. The state of the art does not necessarily imply the most technologically advanced solution. The state of the art described here is sometimes referred to as the “generally acknowledged state of the art”.
State of the art is not the bleeding edge of technology, but what is commonly practiced, and can be defined in standards. For example, BSI recognized ISO 14971:2019 as the “state of the art” risk management standard for medical device risk management. It defines the state of the art in risk management for medical devices, and companies should replace references to earlier versions or other standards in their risk management processes as appropriate. For product-specific standards and horizontal safety standards, companies should refer to the appropriate standards for their products in the design inputs to the products.
Going Forward
Other confusions that continue until today include companies using one risk chart for all devices, even though the acceptable risk levels for different device classes clearly should be different. The requirement in the standard for definition of risk falls to the risk management plan, which can be different for each device or device family being developed, thus providing a method for differentiating risk acceptability for different types of products from the same manufacturer.
Users of ISO 14971:2019 should also have access to ISO TR 24971:2020. EN ISO 14971:2019 and EN ISO TR 24971:2020 are currently identical to the ISO versions. When the European Commission and the European Standards Bodies, CEN and CENELEC, agree on a process to harmonize standards for the MDR and IVDR, it is expected that an amendment will be issued for ISO 14971:2019 containing correspondence tables between the standard and the regulation. Until then, there are no harmonized medical device or IVD risk management standards for the regulations and it is up to the manufacturer to reach compliance with the regulations. EN ISO 14971:2012 is only harmonized to the directives and may not be used for compliance with EU regulations.
With ISO TR 24971:2020, the technical committee responsible for the medical device risk management standard has finally addressed the confusion around the policy for risk acceptability criteria and the difference between that and the criteria themselves. The guidance contains five pages of discussion in Annex C addressing this topic in great detail. An additional short discussion of the requirement in clause 4.4.5 of this same document may also be useful, but the major elements are in Annex C. This discussion should provide the clarification that the medical device industry needs to finally develop a risk management system that meets this requirement of ISO 14971 that has existed since the first edition was published in 2000.
 About The Author
About The Author
Edwin L. Bills has been a member of ISO TC 210 JWG1 for more than 20 years. This is the ISO group responsible for medical device risk management and the creation and maintenance of ISO 14971:2019, the risk management standard for medical devices, and ISO TR 24971:2020, the accompanying risk management guidance.