
8 Key Changes To Understand In The New European MDR And IVDR
By Marcelo Trevino, independent expert
New MDR and IVDR regulations were approved in March 2017 by the European Council and in April 2017 by the European Parliament. Both regulations entered into force on May 26, 2017; new rules will apply starting May 26, 2020, for MDR, and May 26, 2022, for IVDR.
European Parliament. Both regulations entered into force on May 26, 2017; new rules will apply starting May 26, 2020, for MDR, and May 26, 2022, for IVDR.
The new regulations seek to increase medical device safety and effectiveness in the EU Market while addressing weaknesses revealed in the implementation of Medical Device Directives by several medical device manufacturers. The MDR and IVDR also are a response to technical and scientific developments that are quickly shaping the medical device industry. The regulations feature several significant changes and, while many medical device manufacturers already have started implementation efforts, this article aims to provide a quick summary of the key changes, aiding organizations in their plans to transition to the new regulations:
- Vigilance and Postmarket Surveillance (Articles 84, 85, 86, 87, 88, 93) / EUDAMED Database (Article 33) — New regulations associated with vigilance and postmarket surveillance, affecting national authorities and economic operators, include the establishment of an EU database on Medical Devices. The database will capture registration of devices, accredited Notified Bodies, Certificates, Serious Incidents, Safety and Clinical Performance Reports (SSCP), Periodic Safety Update Reports (PSUR), Surveillance Activities, Clinical Investigation Data, Unique Device Identification Information (UDI), and Single Registration Information to correlate information from manufacturer, authorized representative, and importer.
Postmarket surveillance needs to be brought into a continuous evaluation and improvement cycle that links to continuous reviews of risk management information, as well as to a regular update of a public summary of clinical evaluations, safety, and performance. The
new regulations require plans for postmarket surveillance, reports, safety update reports, systems for management of serious incidents and, trend reports (analyzing negative trends vs. risk management documentation).
- Role of Economic Operators (Articles 10, 11, 13, 14, 30) and Person Responsible for Regulatory Compliance (Article 15) — Economic operators include manufacturers, distributors, importers, suppliers, subcontractors, assemblers, and EU Authorized Representatives, all of whom carry responsibility for conformity to the regulations. The new legislation Articles describe obligations that must be carried, including “what” and “how” — representing a major increase in responsibilities for all stakeholders. In addition, a person responsible for regulatory compliance must be available within the organization, with expertise in medical devices demonstrable through experience or qualification.
Summary of Responsibilities

- Scope and Classification of Products (Article 1, Article 2, Article 22, Article 23, Article 51, Article 52, Annex VIII, IX, X, XVI), Strict Rules for substance-based devices and devices that use hazardous substances (Classification Rule 21 and Annex I) and New rules for software and apps (Classification Rule 11) — While the classification system has been retained, rules have tightened and changed for some products, which will result in some devices being reclassified to higher classes. In addition, some devices that were previously exempt from the regulations are now in the scope of the new legislation.
Devices composed of substances or combinations of substances intended to be introduced into the human body — via a body orifice or applied to the skin — and that are absorbed by or locally dispersed in the human body, will have a different classification depending on different factors.
Finally, requirements associated with software should be carefully evaluated to determine potential new classifications.
- Changes affecting Notified Bodies — Supervision of Notified Bodies will change considerably, starting with the requirement that they apply for a new designation. As a result, it is expected that a significant number of notified bodies may not be re-notified, or may not be notified for the same scope, which could force some medical device manufacturers to change notified bodies.
Unannounced audits of manufacturers and authorized representatives will increase, and audit schedules will have to be provided to national authorities ahead of time.
In addition, Notified Bodies will be required to consult with the European Commission on the adequacy of the NBs’ clinical evaluation and postmarket clinical follow-up plans, prior to the NBs granting certificates for Class III implantable and IIb devices intended to administer or remove medicines. Surveillance assessments will include a test of the approved parts and/or materials essential for the integrity of the device, including a check that the quantity of purchased parts corresponds with the quantity of finished devices.
Witness tests and reconciliation activities will take place during on-site audits to ensure that the quality management system (QMS) is working properly.
- Unique Device Identification (UDI)/ Implant Card (Article 27, Article 87, Article 18, Article 19) — UDIs will be introduced to provide traceability on all medical devices and will be placed on the label of the device (not shipping containers). The UDI shall be referenced in Vigilance Reports and will be used for reporting serious incidents and field safety corrective actions. In addition, implant cards with warnings, information about expected device lifetime, and necessary follow-ups — among other requirements — will be required for all implantable medical devices.
- Summary of Safety and Clinical Performance (Article 32) — For implantable devices and for Class III devices, manufacturers must create a summary of safety and clinical performance. The summary must be written in a way that is clear to the intended user — and, if relevant, to the patient — and shall be made available via EUDAMED. Summaries must include, at least, the following:
- Manufacturer Name and Single Registration Number (SRN)
- Device Name and UDI
- Description, previous variant(s), differences, accessories and other products intended to be used in combination
- Intended Purpose, Indications, contraindications and target population
- Possible diagnostic or therapeutic alternatives
- Reference to any harmonized standards and common specifications applied
- Summary of the clinical evaluation report (referred to in Annex IV) and relevant information on post-market clinical follow-up
- Suggested profile and training for users
- Information on residual risks, undesirable effects, warnings and precautions.
- Clinical Evaluation / Postmarket Clinical Follow-up, Clinical Investigations (Article2, Article 55, Article 61, Annex XIV, Annex XV) — Reinforced rules on clinical, performance evaluation, and clinical investigations will require a thorough review of clinical strategy and postmarket clinical follow-up plans. Compliance with current MEDDEVs on clinical requirements will be insufficient, in many cases, to comply with the new rules. Clinical evidence will need to be updated for existing devices, and a summary of the clinical evaluations with convincing, clear evidence will need to be publicly available. For Class III devices and implantable devices, the postmarket clinical follow-up evaluation report, the summary of safety and clinical performance (referred to above), shall be updated at least annually.
Requirements related to clinical data and clinical evaluations are now defined in Article 2, Article 61 and Annex XIV. Clinical investigations are required for Class III and Class IIb implantable devices unless exclusions apply, and justification is provided in accordance to Article 61. Implantable devices intended to remove or administer a medicinal substance will be subject to additional scrutiny, including assessment of the clinical evaluation, instructions for use, and postmarket clinical follow-up plan. For these types of implantable devices, manufacturers may request a consultation from an expert panel prior to its clinical evaluation.
Clinical investigations must be carried out according to the requirements explained in Chapter VI (Articles 61-82); Notified bodies will conduct reviews of the clinical investigation data for conformity to Annex XV.
Postmarket clinical follow-up is a continuous process that updates the clinical evaluation and must be addressed as part of postmarket surveillance plans, looking at data throughout the device’s expected lifetime to help the manufacturer determine whether risks associated with the device remain acceptable.
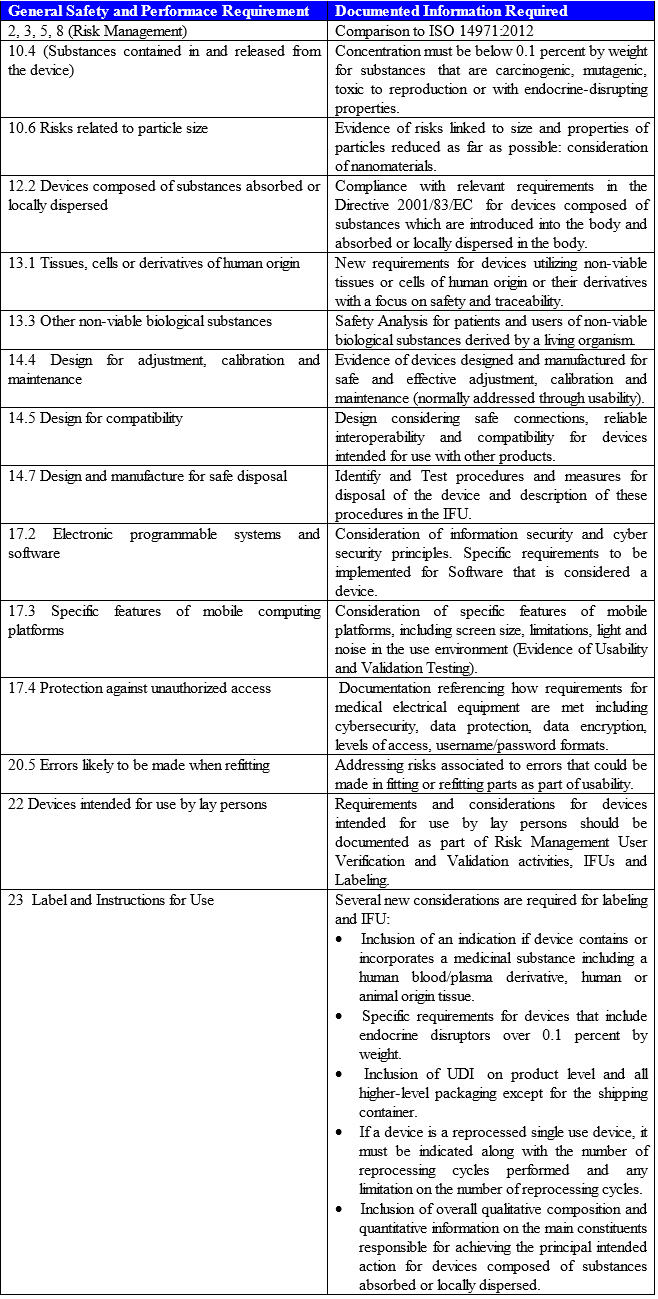
- Summary of Changes in Safety and Performance Requirements (Annex I) — Safety and Performance Requirements replace the Directive’s Essential Requirements. Manufacturers will have to consider how to demonstrate compliance through risk management, testing, technical studies, and other means. Manufacturers then must analyze which requirements are applicable, and document appropriate rationales for requirements that are deemed not applicable. The following table highlights the most important changes:
Developing A Clear Implementation Strategy
While the regulation changes seem overwhelming, defining a good strategy to manage the changes across multiple systems should aid organizations with effective implementation:
- Understand the new legislation and conduct a gap assessment to your current system.
- Develop a detailed plan, considering a cross-functional approach, to determine what other aspects of the quality system will need to be modified.
- Establish separate implementation councils. For example, start with a council that focuses on analyzing product scope and classification, another one addressing the changes to the new QMS, and additional councils to address updated requirements, such as: Safety and Performance Requirements, Clinical Evidence Requirements, and Postmarket Surveillance Requirements. Councils should meet on a regular basis and report progress to senior management until all changes have been implemented. As part of the reporting process, the use of a table or checklist to systematically trace and demonstrate documented compliance with the MDR and the Safety and Performance Requirements will be extremely helpful.
- Consider your QMS interaction with other regulations, and utilize this opportunity to streamline processes while allowing flexibility to incorporate future changes.
- Coordinate expectations and transition plan with your Notified Body.
Conclusion
Despite all the uncertainty with Harmonized Standards and how the MDR actually will be enforced by Notified Bodies and regulators, manufacturers should fully understand the new regulation to prepare for the changes ahead. This includes working closely with their Notified Bodies for consistency in interpretation.
Implementation of UDI, significant changes to technical files and the quality system, and the generation of additional clinical evidence for devices currently on the market are some of the biggest burdens manufacturers will face in the next few years. Therefore, a proactive approach to implement the new regulation — including development of plans for transition and proper allocation of resources — should be prioritized as an important organizational milestone.
About The Author
Marcelo Trevino is the President, Global Regulatory Affairs and Quality Systems, at TregMedical, a life sciences group focused on global medical device regulatory, quality, and compliance. Marcelo can be reached at marcelotrevino@outlook.com
Marcelo has 23+ years’ experience in quality and regulatory affairs, serving in multiple senior leadership roles with different organizations while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, medical imaging/surgical navigation amongst others. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDD/MDR, MDSAP). Mr. Trevino holds a B.S. degree in Industrial and Systems Engineering and an MBA in Supply Chain Management from the W.P. Carey School of Business at Arizona State University. He is also a certified Quality Management Systems Lead Auditor by Exemplar Global.
He has experience working on Lean Six Sigma Projects and many Quality/Regulatory Affairs initiatives in the US and around the world including Third Party Auditing through Notified Bodies, Supplier Audits, Risk Management, Process Validation and remediation activities.
Additionally, he is a Certified Six Sigma Black Belt and Biomedical Auditor through the American Society for Quality (ASQ) and holds Certificates in Environmental & Sustainability Management Regulatory Affairs Management from University of California, Irvine.
He regularly publishes articles to assist corporations in their quest for exceptional quality and regulatory compliance.