
Key Takeaways From The EU MDR Expert Panels' First Published Scientific Opinions
By Muna Kebede and Matthias Fink, AKRA Team GmbH

The Regulation EU 2017/745, the Medical Device Regulation (MDR), which entered into force on May 26, 2021, introduced new requirements for clinical and post-market data for medical devices in Europe. As an additional level of scrutiny for specific devices, the Clinical Evaluation Consultation Procedure (CECP) was implemented in Article 54. The CECP ensures that notified bodies correctly assess the quantity and quality of the clinical data for all class III implantable devices and class IIb active devices intended to administer and/or remove a medicinal product according to Rule 12 of the classification rules in Annex VIII. For these devices, the notified body, after the finalization of the Clinical Evaluation Assessment Report (CEAR), must decide, based on specific criteria, if the CECP is applicable.
Article 52 (2) and the Medical Device Coordination Group (MDCG) guidance document MDCG 2019-3 clarify which class III implantable and class IIb Rule 12 devices do not require a CECP. These three criteria are:
- renewal of a certificate issued under the MDR,
- modification of an already marketed device by the same manufacturer with the same intended purpose without negatively changing the benefit-risk profile, or
- where the notified body confirms compliance with relevant applicable common specification for clinical evaluation of that kind of device.
When the notified body has decided that the CECP is applicable, it will initiate the process via the European Medicines Agency (EMA) as the coordinating authority. There are 10 expert panels1 covering the different medical fields, e.g., cardiovascular, orthopedics, etc.
After a formal check of the submitted documents, the screening panel of the corresponding expert panel will decide within 21 days if a full consultation is required. In cases where a full consultation is required, the expert panel will provide a scientific opinion within 60 days to the notified body. In deciding this, the expert panels will focus on the novelty of the device and significant changes in the benefit-risk profile, or a significantly increased rate of serious incidents reported for a specific group of devices (Annex IX, 5.1 (c) to the MDR).
A manufacturer of a class III implantable or a class IIb Rule 12 device should consider these 60 days for the overall certification process since the notified body cannot continue with the certification procedure before the scientific opinion is provided. If the expert panel delivers a divergent opinion, additional time is required by the notified body and the manufacturer to address these identified differences.
Per Article 52(4), the Commission draws up an annual overview of devices that have been subject to the CECP. This overview also includes all implantable class III and class IIb Rule 12 devices for which the notified bodies decided not to initiate a CECP. The first annual overview, published in January of 2023, covered the period from April 2021 to June 2022.2 During that period, 215 notifications under Article 54(3) were submitted by only eight notified bodies. There are various reasons for the low number of notified bodies that have submitted a notification:
- The designation process of notified bodies is still ongoing, and the cutoff point was in June 2022.
- Not all notified bodies have the scopes for devices requiring a CECP.
Of the 215 submitted notifications, the CECP was requested only in 24 cases. The remaining devices were exempt based on the criteria that these devices were modifications of devices already marketed by the same manufacturer with the same intended purpose. Out of these 24 submissions, 21 were for a class III implantable and three were for class IIb Rule 12 devices. Of these 24 clinical evaluations submitted by the notified bodies as of June 2022, only in six cases the screening panel decided that a full consultation would be required.
In addition to the annual overview, all scientific opinions are publicly available on the Commission´s website.3 Since June 2022, more devices underwent the CECP; by the time of this article being published, 10 scientific opinions have been published by five of the 10 expert panels.
In most cases, the novelty of the device or the related clinical procedure led to the full assessment by the expert panel. In all published opinions, the expert panels did not fully agree with the assessment of the clinical data by the notified body. However, in some cases the panel agreed with the final acceptance of the benefit-risk ratio for placing the device on the market.
The table below presents an overview and the relevant concerns addressed by the panels:
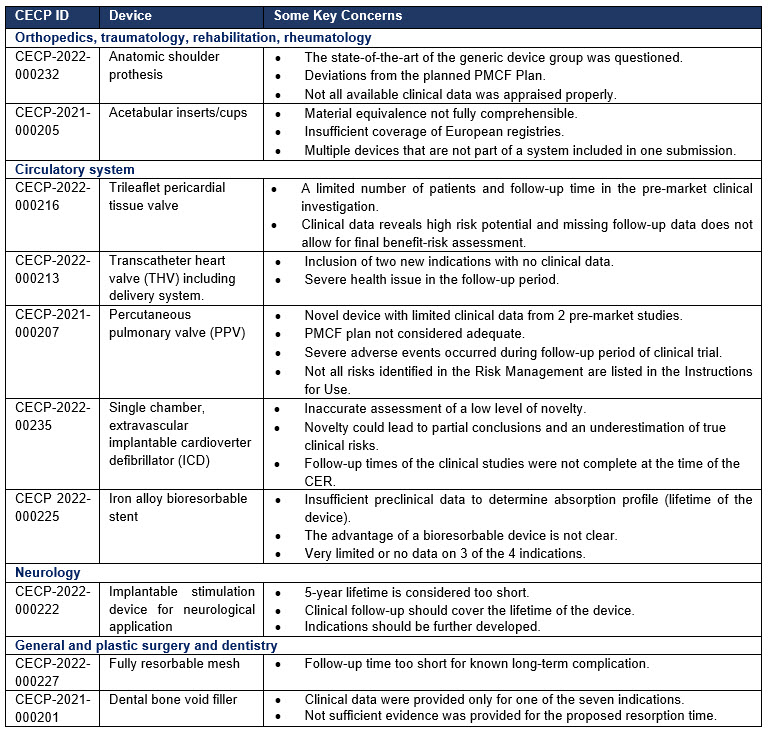
There are certain issues that were addressed in multiple published scientific opinions by different expert panels:
- Concerning the pre-market clinical data: A lack of pre-market clinical data resulted in a challenge of the expected lifetime for a resorbable implant by the expert panel. In one opinion, the study period in the pre-market investigation and the Post-market Clinical Follow-up (PMCF) Plan was not accepted as being sufficient.
- Concerning the state of the art: Manufacturers did not conduct an adequate literature search for the state of the art. Therefore, the overall benefit-risk conclusion could not always be followed by the expert panels.
- Concerning the quality of the clinical evidence: The panels identified several methodological flaws and limitations in the submitted clinical investigation reports. In some cases, the Clinical Evaluation Report (CER) and CEAR did not identify and/or assess existing methodological flaws or limitations. As a consequence, the comprehensibility of the provided benefit-risk conclusion was questioned. Additionally, due to unidentified gaps, the PMCF Plans were not fully accepted.
- Concerning the PMCF: In several opinions, the PMCF Plan was assessed as not sufficient in relation to the available clinical evidence or to the device's lifetime.
Besides their relevance for class III implantable and class IIb Rule 12 devices, the published scientific opinions are an indicator of how certain aspects should be addressed during clinical development and in the documentation. They also can provide valuable insight for devices that do not require a CECP.
Based on the currently available scientific opinions, there are some key takeaways for medical device companies moving forward:
- Identify and include all pertinent favorable and unfavorable clinical data in the clinical evaluation. In some cases, the expert panel might conduct their own literature search.
- Ensure that all components of a system are included in one CER and avoid covering multiple devices not part of the same system in one CER.
- The expert panels focus on the adequacy of the planned PMCF activities especially in relation to the indications and the device lifetime.
- Conduct literature searches, including scientific literature and registries. Appraise them systematically with known and verified methodologies like Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRIMSA) or patient/population, intervention, comparison and outcomes (PICO).
- If you deviate from a study plan of a pre-market clinical investigation plan and submit for CE mark to the notified body before the investigation is concluded, the notified body should challenge this.
The expert panels can be considered the highest level of clinical expertise on the EU level and all relevant stakeholders, like manufacturers and notified bodies, should consider these published scientific opinions for future clinical evaluations and assessments. It remains to be seen if the process will be adapted based on these initial experiences with the mandatory clinical consultations.
References
- https://health.ec.europa.eu/medical-devices-expert-panels/experts/expert-panels_en
- Brussels, 16.1.2023 SWD (2023) 19 final. Annual overview of devices subject to the clinical evaluation consultation procedure pursuant to Article 54(4) of Regulation (EU) 2017/745 on medical devices. https://health.ec.europa.eu/system/files/2023-01/md_annual-overview-cecp_en.pdf
- https://health.ec.europa.eu/medical-devices-expert-panels/experts/list-opinions-provided-under-cecp_en
About The Authors:
 Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
Muna Kebede is a senior consultant at AKRA Team GmbH and supports manufacturers of all device classes to navigate the requirements of the EU MDR. After graduating in human biology & neurobiology, she has gained more than 14 years of experience placing and maintaining medical devices on the market in Europe and globally. In her previous role as director of regulatory affairs and clinical studies, Kebede was responsible for developing and implementing regulatory and clinical strategies for Class III medical devices.
 Matthias Fink, M.D., is a senior clinical consultant with AKRA Team GmbH. He provides consulting for medical device manufacturers of all sizes on regulatory and clinical requirements with a focus on the EU MDR. Previously, he worked as a clinical reviewer and manager of the Clinical Focus Team North America for TÜV SÜD Product Service in Germany and the U.S. A Board-certified orthopedic and trauma surgeon with 17 years of experience in orthopedic, trauma, and reconstructive surgery, as well as cardiovascular and thoracic surgery training, Fink is also a presenter at conferences and workshops on the clinical requirements and the implementation of the EU MDR.
Matthias Fink, M.D., is a senior clinical consultant with AKRA Team GmbH. He provides consulting for medical device manufacturers of all sizes on regulatory and clinical requirements with a focus on the EU MDR. Previously, he worked as a clinical reviewer and manager of the Clinical Focus Team North America for TÜV SÜD Product Service in Germany and the U.S. A Board-certified orthopedic and trauma surgeon with 17 years of experience in orthopedic, trauma, and reconstructive surgery, as well as cardiovascular and thoracic surgery training, Fink is also a presenter at conferences and workshops on the clinical requirements and the implementation of the EU MDR.