
3 Lessons Learned From EU MDR Implementation To Ensure IVDR Adoption
By Hilde Viroux and Maggie Chan (PA Consulting) and Dona O’Neil (Northeastern University)

In the previous article, “How The Right Operating Model For EU MDR Compliance Can Support A Global Footprint,” we discussed key factors for maintaining operational excellence to achieve cost reduction, enhanced compliance, and support global footprint. In this article, we shift our focus toward leveraging lessons learned from EU MDR implementation to ensure seamless transition during IVDR implementation, thereby minimizing the risks of product disruption in the EU market.
The new IVDR requirements present significant challenges for manufacturers, with approximately 90% of all IVD products subject to notified body (NB) review, which is a substantial increase from the current rate of less than 15%. With the limited capacity of NBs, establishing relationships with NBs and engaging in early planning are crucial. These amplify concerns over experience gaps for companies without a quality management system (QMS) for IVDs or any device products, established process and documentation system, or limited experience interacting with NBs. Bridging these gaps and establishing sustainable processes pose additional burdens for IVD manufacturers. However, drawing from EU MDR implementation lessons, IVD manufacturers can navigate this transition, mitigate risks, minimize business disruption, and ensure a smooth transition to IVDR.
For IVD manufacturers, the key lessons learned that can be leveraged from EU MDR implementation include early QMS preparation and planning and collaboration with internal and external stakeholders for project alignment and resource planning, alongside digitalization efforts to support data management and documentation.
1. QMS Preparation And Planning
Manufacturers with an existing QMS for IVDs or medical devices need to conduct a gap assessment against IVDR requirements and develop tailored solutions to bridge the gaps, while those without structured procedures or systems must establish a robust QMS to ensure compliance. ISO 13485 specifies requirements for QMS, and those requirements are harmonized to IVDR. IVD manufacturers are required to comply with the standard. Implementing standardized processes and procedures in accordance with ISO 13485 demonstrates the manufacturer’s commitment to maintaining high standards of quality and compliance. Therefore, a QMS that is developed with insights from subject matter experts in EU MDR can accelerate the approval process with NBs and maintain a competitive position in the IVD industry.
For manufacturers with existing QMS, leveraging the expertise of those who have successfully implemented it, whether it's for IVDR or EU MDR, would be great resources throughout the IVDR implementation journey as there are extensive similarities between EU MDR and IVDR (Table 1). These experts should prioritize IVDR project activities and provide insights into IVDR requirements. Such insights include evaluating the new classification of IVDs, highlighting gaps in existing processes and systems for IVDR, establishing relationships with NBs, and proposing customized solutions to bridge the knowledge gaps.
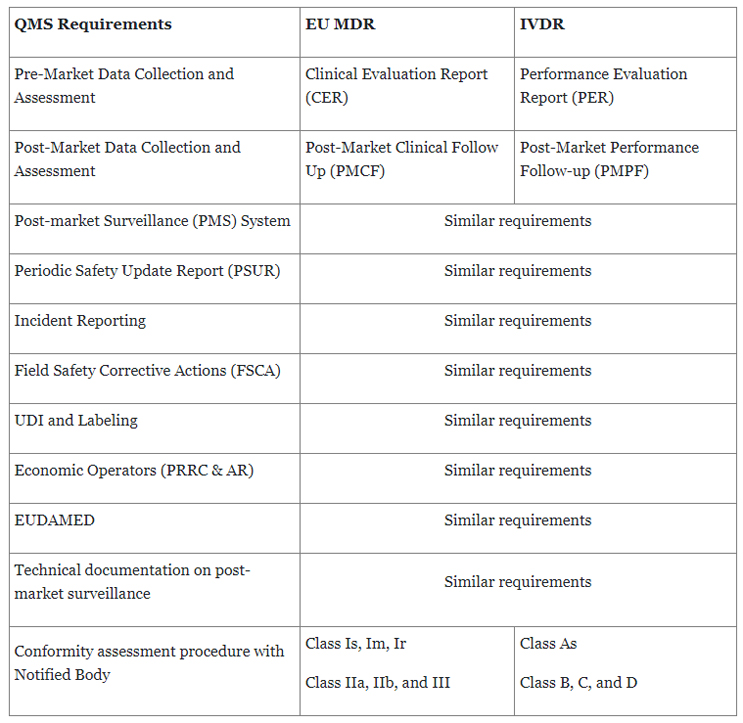
Table 1: Summary of key QMS requirements under EU MDR and IVDR
On the other hand, manufacturers with limited to no experience with QMS or NBs need a dedicated team of subject matter experts (in-house or external) to conduct a gap assessment. The goal of the assessment is to highlight gaps, issues, potential risks, and impacts on the organization against IVDR requirements. This helps create a business case that outlines the requirements to bridge the gaps (knowledge, process, and system), a high-level timeline with key regulatory deadlines, and the associated costs for supporting the IVDR implementation. In addition, this business case should detail potential risks associated with not meeting the requirements by the deadlines, as this is important for senior stakeholders to gain early insight into the process.
2. Collaboration With Internal And External Stakeholders
Furthermore, ascertaining the costs associated with implementing new processes and systems, developing capabilities with the right skillsets, designing training to upskill team members, and selecting new vendors to outsource some of the processes is crucial for the business case before kicking off the implementation project. This information can help senior management to make informed decisions and align the business portfolio with regulatory and market strategies.
Similar to EU MDR implementation, collaboration is the key for a seamless IVDR implementation. It is important to promote cross-functional collaboration and clear communication with stakeholders to ensure everyone understands their roles and responsibilities, milestones are achieved, and compliance is met. A clear road map that outlines timeline, milestones, and key regulatory deadlines is necessary when engaging with stakeholders, not only to ensure alignment but also to enable management to plan their resources accordingly. Furthermore, early engagement with all actors in the supply chain enables a comprehensive understanding of their capabilities in achieving IVDR compliance, so the business case can be prepared accordingly.
IVDR compliance is an ongoing effort, so it is important to emphasize to senior stakeholders the importance of developing a comprehensive strategy for process sustainability and resources management to maintain operational excellence. Prior to the completion of implementation, a new business case should be created outlining the necessary resources (people and technologies) and the associated costs to maintain compliance once the important milestone of IVDR certification is achieved. Without an efficient strategy and comprehensive planning, a manufacturer will soon be at risk of non-compliance and face substantial consequences (e.g., product recalls, warning letters, reputation damage, litigation, fines and penalties, etc.).
Given that ongoing IVDR compliance is a long-term commitment, strategic outsourcing of certain processes to third-party providers may be necessary to ensure efficient resource allocation. Establishing a robust governance structure is crucial to oversee all in-house processes and activities of the supply chain, ensuring alignment with regulatory requirements and organizational objectives.
3. Digitalization
Additional pre- and post-market reporting requirements add burdens, especially for manufacturers without an established QMS or limited experience with NBs. However, digitalization can play an important role in relieving these burdens and streamlining the processes. Digital solutions, such as artificial intelligence (AI) and machine learning (ML) technologies, can significantly enhance efficiency and accuracy by automating routine activities such as data collection, compliance triage, and report drafting. These solutions can streamline the process for drafting various types of regulatory reports, such as clinical performance report, performance evaluation report, technical documentation, and others.
Additionally, a regulatory information management (RIM) system can play a crucial role in managing the submission of regulatory documents to improve traceability. It can facilitate tracking of document signoffs and enable monitoring of document submission locations, ensuring comprehensive traceability throughout the regulatory process. These digital solutions not only enhance data management and documentation efficiency but also streamline the compliance process with IVDR.
Conclusion
For IVD manufacturers, lessons learned from EU MDR implementation offer invaluable guidance when creating the business case for IVDR implementation and post-compliance maintenance, designing an implementation road map, and establishing sustainable processes and systems for IVDR compliance. By incorporating early preparation, implementing collaborative efforts with stakeholders, and utilizing digital solutions for efficient data management and documentation management, manufacturers can mitigate risks, minimize business disruption, and ensure compliance with IVDR requirements. Furthermore, establishing robust governance structures and strategic resource planning is essential for long-term success in this evolving regulatory landscape. Embracing these strategies not only facilitates a smooth transition during IVDR implementation but also fosters a culture of continuous improvement and innovation within the IVD industry.
About the Authors:
 Hilde Viroux is a medtech expert at PA Consulting and is an expert on the European Medical Devices Regulation. She has broad experience in regulations, quality, manufacturing, supply chain, and project management in the pharmaceutical and medical device industry. She has an MSc in medical technology regulatory affairs from Cranfield University in the U.K. and a BS in biochemistry engineering.
Hilde Viroux is a medtech expert at PA Consulting and is an expert on the European Medical Devices Regulation. She has broad experience in regulations, quality, manufacturing, supply chain, and project management in the pharmaceutical and medical device industry. She has an MSc in medical technology regulatory affairs from Cranfield University in the U.K. and a BS in biochemistry engineering.
 Maggie Chan is a life sciences and regulatory expert at PA Consulting. She focuses on leading process and operation improvement for medical device and pharmaceutical companies. She has led labeling remediation and e-labeling process design projects in compliance with global medical devices regulation for both implantable and non-implantable medical devices. She has been supporting companies by building capabilities and designing processes to ensure they comply with ISO 13485, EU MDR, 21 CFR, etc., in line with the product portfolio. She has a Master of Science in law from Northwestern Pritzker School of Law in the U.S. and a BS in biology.
Maggie Chan is a life sciences and regulatory expert at PA Consulting. She focuses on leading process and operation improvement for medical device and pharmaceutical companies. She has led labeling remediation and e-labeling process design projects in compliance with global medical devices regulation for both implantable and non-implantable medical devices. She has been supporting companies by building capabilities and designing processes to ensure they comply with ISO 13485, EU MDR, 21 CFR, etc., in line with the product portfolio. She has a Master of Science in law from Northwestern Pritzker School of Law in the U.S. and a BS in biology.
 Dona O’Neil is an industry EU MDR expert and an adjunct professor for Northeastern University’s master’s program in regulatory affairs. She has experience developing and implementing EU MDR clinical evidence requirements across many therapeutic areas and all device classifications. O’Neil has an MPH (healthy policy concentration) from George Washington University and is certified as a clinical research professional by SOCRA.
Dona O’Neil is an industry EU MDR expert and an adjunct professor for Northeastern University’s master’s program in regulatory affairs. She has experience developing and implementing EU MDR clinical evidence requirements across many therapeutic areas and all device classifications. O’Neil has an MPH (healthy policy concentration) from George Washington University and is certified as a clinical research professional by SOCRA.