
Long Live Design Controls: Navigating The Shift From QSR To QMSR
By Jayet Moon and Arun Mathew

In the realm of medical device development and manufacturing, adherence to stringent regulatory standards is not just a legal requirement, it’s also a fundamental aspect of ensuring product safety and efficacy. For years, the FDA's design controls under 21 CFR Part 820 (Quality Systems Regulation [QSR]) have served as a cornerstone for guiding manufacturers through the process of designing and design transfer of medical devices. However, a recent shift in regulatory focus toward a harmonized global approach has led to significant changes, notably with the adoption of the Quality Management System Regulation (QMSR) that goes live on Feb. 2, 2026. This transition has effectively superseded the explicit design controls CFRs and associated FDA guidance, but their essence lives on within the framework of ISO 13485:2016 Clause 7.3.
ISO 13485, the internationally recognized standard for quality management systems in the medical device industry, plays a pivotal role in this regulatory evolution. While the explicit design controls CFRs have been removed in QMSR, the spirit of design controls remains deeply ingrained within ISO 13485. The standard emphasizes the importance of a systematic approach to product development, encompassing design and development planning (Clause 7.3.2), design inputs (Clause 7.3.3), design outputs (Clause 7.3.4), design review (Clause 7.3.5), verification (Clause 7.3.6) and validation (Clause 7.3.7), design transfer (Clause 7.3.8), control of design and development changes (Clause 7.3.9), and design and development files (Clause 7.3.10).
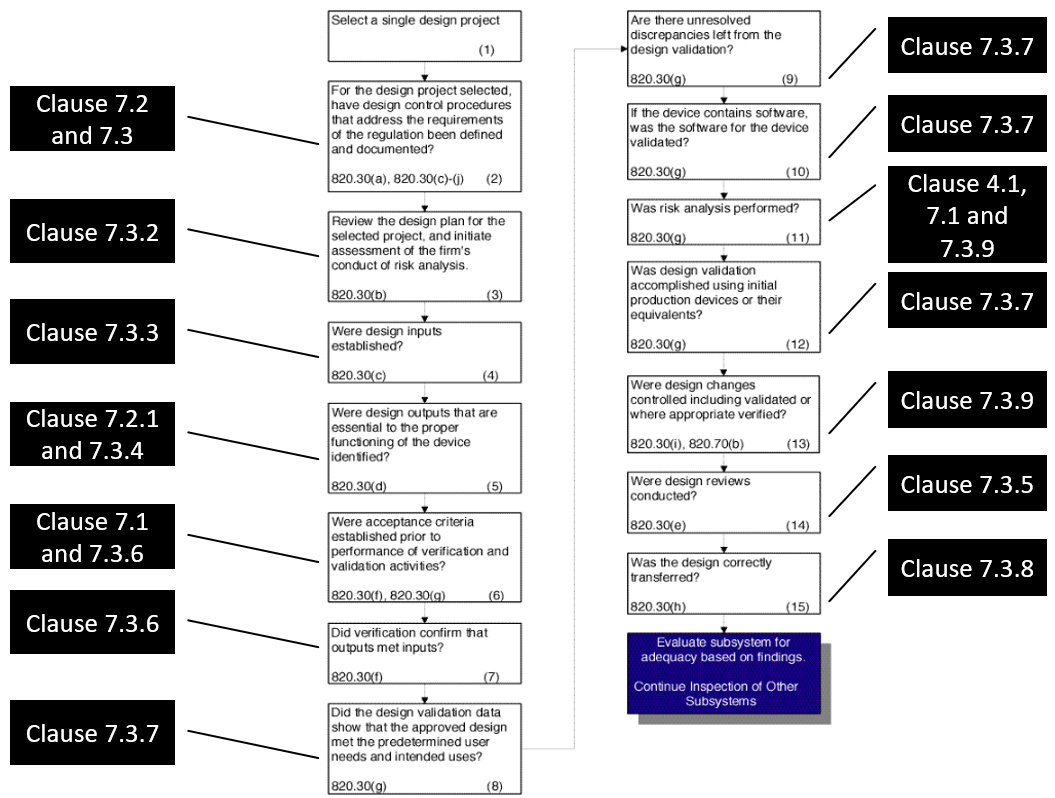
Figure 1: QSR to ISO 13485:2016 for design controls (Note: 7.3.3 also applies to 820.30(g); 820.30(j) is missing above and should correspond to 7.3.10). Click on image to enlarge.
A great document where FDA’s approach to design controls is presented succinctly is the Guide to Inspection of Quality Systems released by FDA in 1999. While this guide will be updated to match QMSR requirements in the near future, we have associated the relevant clauses from ISO 13485 to the old CFRs from QSR in Figure 1 to show how FDA can still follow the same inspection path using ISO 13485 clauses. In the MDSAP audit approach, where FDA is a key member with regard to chapter 5 on design and development, the references to the relevant ISO 13485 clauses are provided. This audit approach also discusses the design controls.
This shows that while the letter of the law may change, the underlying principles and objectives remain constant, at least in this specific case. Manufacturers must recognize that adherence to design controls is not merely a checkbox exercise but a commitment to ensuring the safety, effectiveness, and quality of medical devices. The traditional waterfall approach for product design controls as shown in Figure 2 and reproduced from the 1997 Design Control Guidance for Medical Device Manufacturers is still fully compatible with ISO 13485:2016, and all this old guidance will still apply when the CFRs are replaced with their ISO equivalent clauses. Of course, design history files per old 820.30(j) will now be known as part of design and development files per Clause 7.3.10. It is to be seen if and how FDA will incorporate Agile and V model methods of product development in new guidance(s).
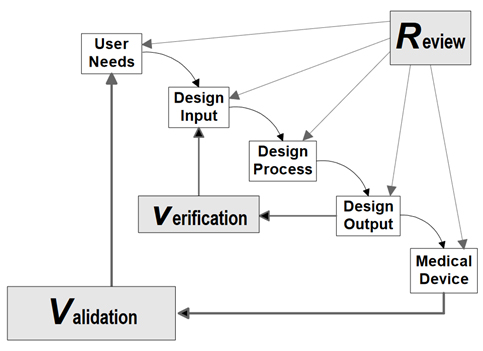
Figure 2: Application of Design Controls to Waterfall Design Process. Reproduced from the 1997 Design Control Guidance For Medical Device Manufacturers.
A key change in the new QMSR era will be the stringency with which FDA will enforce the varied risk management clauses in 13485 that rely heavily on ISO 14971 as part of design and development .ISO 13485 requires organization to have one or more risk management processes for risk management in product realization and, per the note in the standard, it refers to ISO 14971. ISO 13485 requires that outputs of risk management activities be inputs to the design and development process. Not only that, but it also requires implicit adherence to risk management process per ISO 14971 throughout design/product realization processes and in purchasing and manufacturing processes as appropriate. On a higher level, ISO 13845 requires organizations to apply a risk-based approach to the control of the appropriate processes needed for the quality management system.
Organizations should manage this transition by:
- Education and Training: Ensure that personnel are adequately trained in the principles of ISO 13485 and understand how they align with traditional design controls and risk management requirements.
- Documentation: Update quality management systems documentation to reflect the shift toward QMSR and incorporate relevant clauses from ISO 13485 pertaining to design and associated documentation. Pay special attention to Clause 4.2 and 7.3.10.
- Risk Management: Place a renewed emphasis on risk management throughout the product life cycle, aligning with the requirements of ISO 14971 and integrating risk considerations into design and development activities. Risk management goes beyond the design phases into the production and post-production phases as well.
- Continuous Improvement: Embrace a quality culture of continuous improvement, leveraging feedback from post-market surveillance, customer complaints, and other sources to refine design and development processes.
- Internal Audit: Referencing the ISO 13485 requirements ensuring the risk-based approaches are followed in the quality management systems and in design control concepts.
The transition from FDA design controls CFRs to QMSR represents a significant milestone in the evolution of medical device regulation, signaling a shift toward global harmonization and alignment with international standards. While the explicit regulations may change, the underlying principles of design controls endure, serving as a guiding light for manufacturers committed to ensuring the safety, effectiveness, and quality of medical devices. By embracing this shift and leveraging the framework provided by ISO 13485 and 14971, organizations can navigate the changing regulatory landscape with confidence, secure in the knowledge that the spirit of design controls lives on, even if the word itself may not.
In part two of this article series, we will explore further integration of design and development process and risk management in greater detail.
About The Authors:
 Jayet Moon earned a master’s degree in biomedical engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI)-Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS)-Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Foundations of Quality Risk Management, was recently released by ASQ Quality Press. He holds ASQ CQE, CQSP, and CQIA certifications.
Jayet Moon earned a master’s degree in biomedical engineering from Drexel University in Philadelphia and is a Project Management Institute (PMI)-Certified Risk Management Professional (PMI-RMP). Jayet is also a Chartered Quality Professional in the UK (CQP-MCQI). He is also an Enterprise Risk Management Certified Professional (ERMCP) and a Risk Management Society (RIMS)-Certified Risk Management Professional (RIMS-CRMP). He is a Fellow of the International Institute of Risk & Safety Management. His new book, Foundations of Quality Risk Management, was recently released by ASQ Quality Press. He holds ASQ CQE, CQSP, and CQIA certifications.
 Arun Mathew works in quality systems in R&D at AbbVie. He holds an executive MBA, M.Sc., in real time embedded systems, and a diploma in electronics. He has more than two decades of experience in the medical devices industry, drug product development, manufacturing, and regulatory affairs. He is a longtime member of the American Society for Quality and is a certified CQA Certified Quality Auditor and CQE Certified Quality Engineer. Previously, Mathew has worked at Fortune 500 companies such as Zimmer Biomet, Medtronic, and Baxter.
Arun Mathew works in quality systems in R&D at AbbVie. He holds an executive MBA, M.Sc., in real time embedded systems, and a diploma in electronics. He has more than two decades of experience in the medical devices industry, drug product development, manufacturing, and regulatory affairs. He is a longtime member of the American Society for Quality and is a certified CQA Certified Quality Auditor and CQE Certified Quality Engineer. Previously, Mathew has worked at Fortune 500 companies such as Zimmer Biomet, Medtronic, and Baxter.