
Medical Devices Regulatory Priorities In India
By Harish Reddy Arkala, Freyr

The Indian device market is among the top 20 across the globe and is the fourth-largest market in Asia. Its market value is approximately USD $5.5 billion, with an expected growth rate of 15 percent CAGR.
This market has witnessed continuous transformation over the past two decades. Before the New Economic Policy (1991), it was dominant in domestic manufacturing circles. Later, it transformed into an import-driven market. Prior to 2006, the medical device sector in India was unregulated; that era ended in 2006, when the Central Drugs Standard Control Organization (CDSCO) notified 15 medical devices for which registration is required.
In solidarity with the Make in India program, the CDSCO published the new Medical Device Rules, 2017, which came into force on Jan. 1, 2018. Prior to implementation of the Medical Device Rules, 2017, notified medical devices were regulated as drugs (pharmaceutical products) in India under the Drug and Cosmetic Act, 1940. Therefore, it was required to distinguish medical devices from pharmaceutical products. Secondly, there was an urgent need to provide a more conducive environment for local manufacturers to set up industries in India. Finally, the Ministry of Commerce and Industry issued a public procurement in 2017, and identified the Department of Pharmaceuticals as a Notified agency.
The new rules have been formulated to promote domestic manufacturing and to regulate import and manufacturing in the region. Currently, multinational companies occupy approximately 75 percent of sales in the Indian medical device market. The new regulations follow the GHTF (Global Harmonisation Task Force) guidelines and are in consonance with these rules’ risk-based classification. In addition, inspections by notified bodies have been introduced in the new medical devices rules. In this article, we highlight some of the key points for a better understanding of the Medical Device Rules, 2017.
Classification
In consonance with global regulations, the new rules introduced a risk-based classification system. The CDSCO classifies these devices and publishes the list of classified devices from time to time on its website. Importers and manufacturers are required to follow the classification list to classify their devices. If the classification is higher in GHFT countries, then a higher grade of classification will be considered.
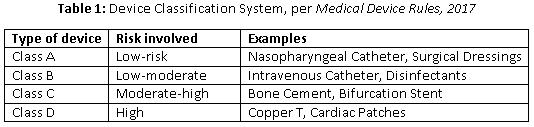
Quality Management System (QMS) Assessment
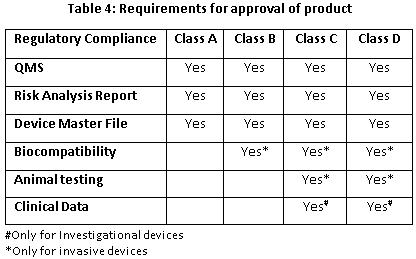
Per the new rules, a new procedure of “third party conformity assessment and certification’” through Notified Bodies has been introduced. The notified body can perform a QMS assessment at manufacturing sites for Class A and Class B devices. Upon request, the notified body also can support CDSCO for Class C and Class D medical devices’ manufacturing site QMS assessments. The accredited list of notified bodies then will be displayed by CDSCO on its website. In case of foreign manufacturers, CDSCO may also require an inspection of the overseas manufacturing site, either by in-house CDSCO inspectors or by any notified body.
Registration
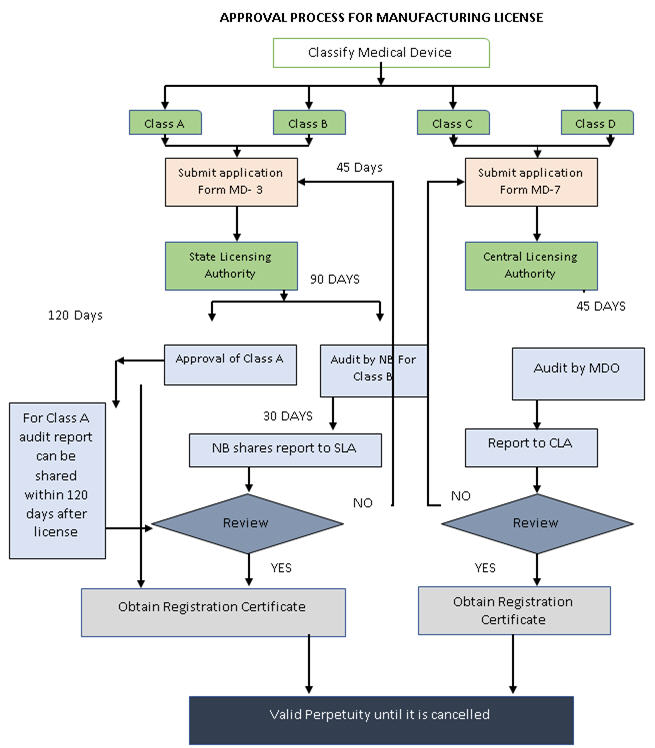
The new rule is going to make it compulsory to obtain manufacturing and import licenses for all devices. All the applications for manufacturing and import licenses are processed through an online portal, SUGAM — an online licensing system that belongs to the Ministry of Health and Family Welfare.
The State Licensing Authority (SLA) will regulate the Manufacturing License for Class A and Class B devices, whereas Class C and Class D license application will be presented to the Central Licensing Authority (FSSAI). A Quality Assessment Report (QAR) must be submitted for Class B, Class C, and Class D devices, along with an application for a manufacturing license. In contrast, a QAR for Class A medical device needs to be submitted within 120 days from the date of grant of Manufacturing License.
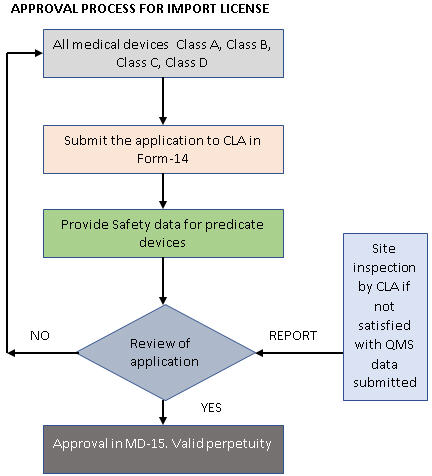
In case of an Import License, a license for manufacturing or distribution is a prerequisite. Foreign manufacturers should appoint an authorized Indian agent to hold the license and carry out post marketing surveillance (PMS) activities, as well as distribution of medical devices. Import Licence applications for all classes of medical devices are to be presented to the Central Licensing Authority.
In contrast to the Manufacturing Licence, an Import License does not require a QAR, but the Central Licensing Authority may inspect the foreign premises, if required. To make the new guideline even more stringent, it now is mandatory to submit the complete Technical File and Import License application to the Central Licensing Authority, and every Indian agent will be responsible for the PMS activities within the country. Multiple Import Licenses for the same product, by different Indian agents, are possible under the 2017 rules.
All licenses granted are perpetual unless they are cancelled. In order to retain the license, one is required to pay license retention fee every five years. The Indian government has made special note of the inevitability of timelines, and rationalized the time required to grant a license.

Clinical Investigation
The Medical Device Rules, 2017 changed the clinical trial scenario for an investigational medical device from a four-phase trial — like those for drugs, per Schedule Y — to a two-phase trial. The two phases will be divided into pilot clinical investigation (Exploratory Study) and pivotal clinical investigation (Confirmatory Study). In addition to this, PMS also is compulsory after gaining marketing approval for the device. However, for an Import License of a medical device without any predicate device in India, clinical investigation would not be required if a Free Sale Certificate (FSC) has been issued by a competent authority from Australia, Canada, Japan, the United States, or member states of European Union.
Additionally, “Substantial Equivalence” to a predicate device for medical devices (except investigational medical devices) has been introduced in the new rules. In case of in-vitro diagnostics, “Clinical Performance Evaluation” will be part of the regulatory requirement. Further, no prior approval of a clinical trial is required for academic clinical trials, provided the data generated is not used for obtaining manufacturing and import license.
Labeling
It is mandatory to follow labelling requirements according to specifications outlined in the new rules. In addition to this, the Indian government also has made it mandatory to follow the Legal Metrology (Packaged Commodities) Rules, 2011. Moreover, the Unique Device Identification (UDI) number for each device must be mentioned on the label, effective January 2022.
Recalls
The Drug and Cosmetic Act fails to obligate the manufacturer or importer to withdraw the product from the market. Per the new rules, the manufacturer or importer must recall any product that is dangerous or harmful, and they are required to provide a reason for the recall.
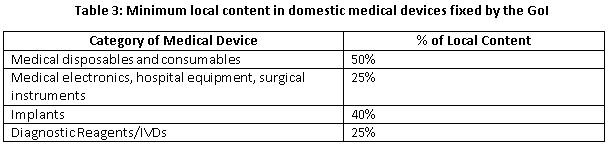
Public Procurement Order (PPO)
To improve ecosystem for domestic manufacturers, Government of India (GoI) brought draft guidelines for PPO. These rules cover tenders valued at 50 lakhs or less for the public procurement of medical devices. The determination of local content cost shall be based on manpower and country of origin of material (direct component). The Government proposed a formula to calculate local content:
D= (A/C)*100
(C=A+B)
D= Percentage of Local Content
C= Total Cost
B= Cost of Imported component
A= Cost of Domestic component
Conclusion
The Medical Device Rules, 2017 have many attractive features that encourage the medical device sector in India. By introducing a single online portal, the registration process has been streamlined. An audit by the notified bodies will further increase the manufacturing quality of devices. A change in clinical trial requirements will encourage the innovation of new medical devices. The regulations will thus encourage domestic manufacturing and increased scrutiny of Import License documents.
About The Author
Harish Reddy Arkala is an Associate in the Medical Devices branch of Freyr’s Strategic Services. Harish completed his major in Drug Regulatory Affairs from Amity University. Harish is an expert on Indian drug and medical device regulations at Freyr. He has a keen interest in tracking new regulations pertaining to India, and their implementation, to analyze how they impact business factors in the current scenarios.