
2 Of The Most Common IVDR Challenges (With Solutions)
By Eljar Amini-Nejad, NSF Health Sciences

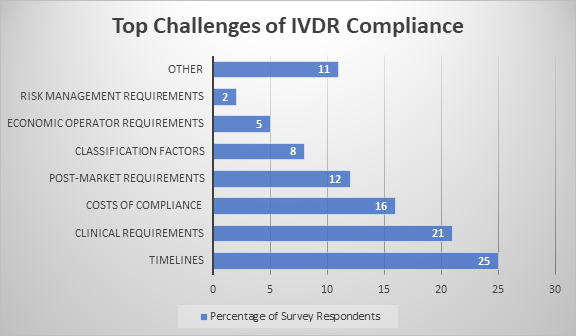
As the May 26, 2022, deadline for the new In Vitro Diagnostic Medical Device Regulation (IVDR) (EU) 2017/746 gets closer by the day, in vitro diagnostic (IVD) manufacturers and notified bodies across the industry continue preparing for the transition. Although many have made significant progress over the last year, lingering doubts and challenges remain. A recent survey co-sponsored by NSF International found that clinical and postmarket requirements are among the most significant challenges in the regulation, with approximately one-third (33%) of survey respondents citing clinical (performance evaluation) requirements or postmarket requirements as the most significant challenges.
Under the IVD Directive 98/79/EC (IVDD), as many as 80% of IVDs were permitted to self-declare that a product conformed to the directive. The IVDR requires that medium- to high-risk IVDs must undergo an assessment of conformity by a notified body. The IVDR, with its new concept of classifications, effectively flips the landscape, leaving an estimated 80% or more IVDs in need of a notified body assessment.
The COVID-19 pandemic added additional challenges as a significant reduction in staffing resources spread across nearly every industry, including portions of healthcare, leaving many concerned about the timeline for compliance. Today, as staffing returns to pre-pandemic levels and on-site work resumes, IVD manufacturers and others in the industry must resume typical daily operations, catch up on work delayed due to the pandemic, and continue working toward IVDR compliance while remaining competitive and innovative in the market. Those who saved much of this process for this year undoubtedly face a more difficult road ahead due to timing, but the complexities of both performance evaluation and postmarket requirements are felt across the industry.
However, despite these valid concerns, timely compliance remains possible. With a more comprehensive understanding of the new regulation and proper preparation, both performance evaluation and postmarketing requirements can be satisfied to help IVDs remain on the EU market. It is time to bring your technical documentation into compliance with the IVDR.
Changes To Performance Evaluation Requirements
The clinical-related performance evaluation requirements under the new regulations are complex and may seem overwhelming to some. In the recent survey, only 16% of organizations expressed full comprehension of what is required to comply with these requirements. Under the old directive, manufacturers were required to supply evidence at various levels of breadth and depth, but the new regulation details comprehensive requirements that are also prescriptive. Along with demonstrating clinical and analytical performance as well as scientific validity, manufacturers must demonstrate the clinical benefits of the IVD and why it can be considered state of the art.
Needs will vary based on current preparedness and the product itself. To better understand what is necessary for compliance, manufacturers must reevaluate their existing evidence and perform a gap analysis. From there, new evidence must be generated to address any identified gaps, and the work required for this process should not be underestimated. Though the risk class of a device will influence the complexity of clinical and analytical performance as well as scientific validity, this is only one factor.
Previously non-listed devices under the directive will be challenged with providing evidence requirements at an unprecedented scale to prove safety and performance of the IVD. Devices already regulated under the directive will face new and more detailed requirements under the regulation, and evidence gathering should begin quickly if efforts are not already underway.
Fortunately, the new requirements are not entirely rigid. Both real-word data and evidence from peer-reviewed literature are permitted with adequate justification. This literature can help provide performance evaluation information while also supporting scientific validity and demonstrating the state-of-the-art qualities of the device.
Following compliance with IVDR, a continuous evaluation of performance remains necessary. Postmarket surveillance (PMS) plans must address the creation of a postmarket performance follow-up (PMPF) plan. This helps guide you through the regular updating of your performance evaluation, eliminating any postmarketing confusion.
Postmarket Requirements
Though a vast majority of organizations appear to have a sound understanding of postmarket requirements, over half of these organizations (54%) say they are only somewhat prepared. Some of this may be due to misconceptions surrounding requirements for devices of different classes; Chapter VII and Annex III in the new IVDR dictate postmarket surveillance requirements for devices at every risk level. The manufacturer’s documentation on postmarket surveillance is based on the PMS plan, outlined in Section 1 of Annex III. It must allow for the systematic gathering and analyzing of relevant data related to quality, performance, and safety. This data demonstrates continuously maintained conformity, ensures missing clinical data is captured, and helps manufacturers initiate preventive and corrective actions.
However, manufacturers may have more in place than they are aware of. If a manufacturer’s quality management system (QMS) already includes a built-in postmarket surveillance process (see Article 10(8i)), postmarket surveillance planning can begin. When this postmarket surveillance is paired with an understanding of the device itself — including risk assessment outputs, novelty, and complexity — the process helps establish the PMS plan.
Table I: Postmarket Surveillance Requirements by Class. Different postmarket surveillance requirements exist based on device class, according to Article 80 and 81.

In all cases, remember that your technical documentation must be presented in a clear, organized, readily searchable, and unambiguous manner and address all the elements listed in Annexes II and III. Ensuring this level of organization will also streamline efforts as you prepare for certification and can help ease the process of meeting all other requirements, including postmarketing.
In summary, if either performance evaluation or postmarket needs are left until later or assumed to be non-issues due to previous compliance with the IVDD, timelines will likely only become more concerning. Both sets of requirements necessitate planning, due diligence, and proper execution to meet the current timeline. Further, with notified bodies currently facing potential IVDR-related capacity issues, waiting may lead to more extensive delays and, as a result, removal from the market until registration is earned.
 About The Author:
About The Author:
Eljar Amini-Nejad is a senior consultant at NSF Health Sciences in Hamburg. He specializes in medical device and IVD quality management and regulatory affairs consulting.