
Planned CDRH Reorganization Will Impact Device Approvals and Inspections
By Jerry Chapman

The FDA Center for Devices and Radiological Health (CDRH) has proposed a reorganization that will collapse the Center’s seven offices into four, combining siloes of expertise into a new office that aligns talent into teams by product type and technology. Individuals from the evaluation and compliance groups for specific product types and technologies will be on one team, creating a new review and inspection dynamic.
The proposed CDRH Office of Product Evaluation and Quality (OPEQ) will comprise several Office of Health Technology (OHT) teams. Each technology-specific team will focus combined expertise from the evaluation, compliance, surveillance, and radiological health offices to streamline the performance of regulatory reviews, as well as improve communications.
At a Xavier Health web conference in February, Half Day with FDA — The Future: Alignment and Reliance, CDRH Office of Compliance Regulatory Affairs Deputy Director Capt. Sean Boyd presented the reorganization plan and the reasons for its proposal. The plan has been submitted for agency review, and its approval and implementation is anticipated by the end of 2018.
In Boyd’s talk, Total Product Lifecycle (TPLC) Realignment within CDRH, he characterized the proposed reorganization as “a new way of doing business that allows us to better focus on the total product lifecycle of devices, manufacturers, and the medical device industry.”
He explained that the effort began a decade ago with CDRH’s Office of In vitro Diagnostics (OID). That office, employing a similar reorganization model as the current proposal, evolved to become the Office of In vitro Diagnostics and Radiological Health (OIR) about five years ago.
Breaking Down Silos
In the current CDRH structure, Boyd commented, “these large offices – which may be between 200 and 600 people – naturally create barriers within the Center that can hamper sharing information across the Center, and hamper making decisions that are informed by everything that we know in the timeline in which we would want to make those decisions.”
Primarily impacted by the reorganization will be the:
- Office of Device Evaluation (ODE)
- Office of Compliance (OC)
- Office of Surveillance and Biometrics (OSB)
- Office of In-vitro Diagnostics and Radiological Health (OIR)
CDRH intends to combine staff and managers from these offices, as well as the functions they perform, into “an entirely new organization,” said Boyd. He explained that the Offices of Science and Engineering Laboratories (OSEL), Communication and Education (OCE), and Management (OM) will not be impacted by the restructuring, “but will be impacted in terms of the connections that we need to maintain or refine across the new organization.”
The intent is to “eliminate silo structures, bring people together so that they are aware of all the information the Center has, and enable us to make better decisions around the specific technologies that we review.”
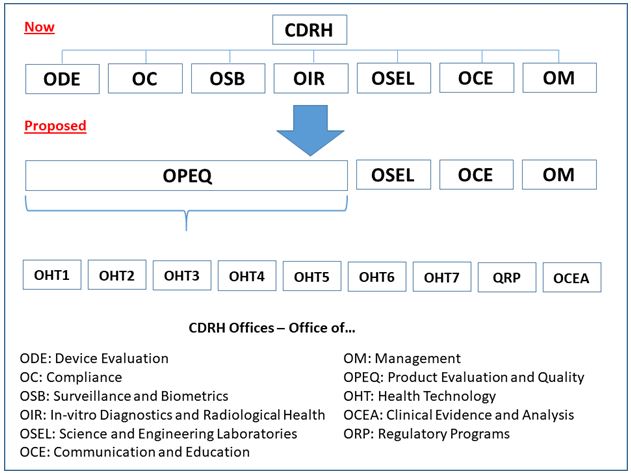
New Offices of Health Technology Combine Expertise
Within OPEQ, there will be seven Offices of Health Technology (OHTs) “that are aligned in much the way that our pre-market divisions are organized in ODE today,” Boyd continued. Each office will be responsible for pre-market review, compliance and enforcement, and surveillance functions for specific types of medical devices.
For example, people who today handle pre-market reviews on cardiovascular devices, compliance and enforcement reviews related to cardiovascular devices, or surveillance related to cardiovascular devices, all will be combined into one OHT under the new organizational structure — likewise for other device areas, or groupings of device areas.
Within each OHT, there will be further breakdowns along more specific technology lines. Also, in addition to individual expertise in various functions, each team may include people familiar with all of those functions — for example, a generalized reviewer who can operate in all of those functional areas.
“Importantly,” the CDRH official stressed, “Those teams are going to be sharing information and interacting with one another on a regular basis to share what they know about something they have seen — for example, in a pre-market file that is in-house, or in a recall, or in an inspection report review, or something that they have seen in a series of adverse event reports that they have looked at, or another signal that they are looking at. People are then able to better communicate the things that they know that might inform questions that we ask during one of our reviews.”
Also, the information sharing “might inform a path that we take to either achieve compliance or interact with a firm to better understand a technology or what is going on with a manufacturing process. So, having people who share that information and collaboratively work to reach those decisions, we think, is a better model.”
In addition to the OHTs, OPEQ will contain two offices charged with management of the regulatory programs: the Office of Regulatory Programs (ORP) and the Office of Clinical Evidence and Analysis (OCEA). These offices will be responsible for development of policies, programs, and processes implemented within the OHTs.
“One of the primary goals of these supporting organizations,” Boyd emphasized, “will be to define the policies and processes, and deliver the tools and templates that promote consistent application of policy across the Center.” The intent is to make decisions founded on the regulations and requirements, and to ensure “that we are modifying our approaches appropriately and consistently across all of those offices.”
One-Stop Shop Envisioned
Boyd said he does not expect to see immediate effects following the reorganization but, over time, the vision is for the OHTs to provide device manufacturers with a “one-stop shop” for regulatory and compliance information.
The combined expertise will be “all on one team, one division, or one office, which will be able to address whatever needs you have. Teams can paint the full picture of what is going on with a particular device or firm.” As a result, he added, industry should experience “better, more streamlined interactions, and better informed decisions or outcomes.”
This will result from CDRH having a better understanding of firms’ operations and being more familiar with how the various organizations function — for example, Boyd noted, “where you excel, where you are growing, and where there may be issues that we might need to work together to address.”
The one-stop shop will be enabled by OHT teams:
- having improved knowledge of the technologies that firms are bringing to market
- having concentrated sets of individuals, focused on particular subsets of each technology
- understanding, from a pre-market review perspective, what development, design, performance evaluation, and testing has been completed
- understanding some of the clinical assessments performed to prove that the device is ready to come to market.
Both Small And Large Firms Will Benefit
At the end of Boyd’s presentation, Xavier Health director and session moderator Marla Phillips commented that the one-stop shop goal “is fantastic, especially if you are a small company and you have one or two products that are typically within the same type of technology category. You are going to get to know your team in CDRH very well, and have a great interaction.”
She also noted that “larger companies have multiple technologies, but often have teams within the company that are working based on specific types of technologies, so they will have a specific team that they are going to get to know well.”
Regarding those larger companies with multiple technologies — and thus interactions with multiple OHT teams, Phillips asked how CDRH will ensure that the various teams communicate openly with each other regarding that company: “How will they have a wider company view when they are working in these more silo approaches?”
The plan, Boyd responded, is to have “‘communities of practice’ in place that are designed to ensure that we are taking a holistic approach across multiple OHTs – for example, for a specific area that might be related to pediatrics or women’s health or some other cross-cutting or common area that could affect a single manufacturer who is operating in different device areas. We might have networks of individuals who work across multiple OHTs, who are familiar with some of the larger manufacturers that work in different device technology areas, who are routinely coming together to share what they know about a particular firm.”
Boyd also noted that the Office of Regulatory Programs will have the responsibility “to look at data that we have [internal], or external sources of data, to identify trends that we are seeing across multiple OHTs that might be industry wide. We will look for trends that we are seeing for a particular manufacturer that works across multiple technology areas and share that information with the OHTs so that we are making the connections that we need to in our interactions with that firm on multiple different fronts.”
Total Product Lifecycle Database is Available
The publicly available CDRH Total Product Life Cycle (TPLC) database integrates premarket and post-market data about medical devices. It includes information pulled from CDRH databases, including Premarket Approvals (PMA), Premarket Notifications (510[k]), Adverse Events, and Recalls. The
TPLC database is refreshed as each of the individual data sources is updated, and it can be searched by device name or product code to receive a full report about a particular product line.
In its current form, the TPLC database provides data by product code (procode), or generic category of device, and not by individual submission or brand name. A list of information sources that feed into the database is available here.
About The Author
Jerry Chapman is a GMP consultant with nearly 40 years of experience in the pharmaceutical industry. His experience includes numerous positions in development, manufacturing, and quality, at the plant, site, and corporate levels. He designed and implemented a comprehensive “GMP Intelligence” process at Eli Lilly and again as a consultant at a top-five animal health firm. Jerry served as Senior Editor at International Pharmaceutical Quality (IPQ) for six years, producing in-depth reports of interest to industry and regulators in the drug GMP/CMC space. Email Jerry here, visit his website here, or connect with him on LinkedIn here.