
Seeking A Smooth Transition To The New EU MDR? (Then Start Complying Now)
By Tido Eger, Navigant
The new Medical Device Regulations (MDR) were published in the Official Journal of the European Union on May 5, 2017 [1]. From that date, manufacturers, suppliers, Notified Bodies, and national competent authorities have a transition period of three years to comply with the new set of rules. Given the large scale of changes, there is great pressure on all actors to analyse the MDR, conduct impact assessments, and implement compliant processes. After May 2020, non-compliance threatens CE-mark certification, access to the European market, or, in the case of Notified Bodies, re-designation.
The new regulation has a word count that is nearly four times higher than its predecessor, the Medical Devices Directive (MDD) [2], contains five more annexes, and uses the word “safety” 290 times, rather than just 40 times. This paper focuses on five topics relevant to the upcoming changes:
- Implications for Notified Bodies, and for organizations working with Notified Bodies
- Impact on product portfolios and classification
- The need for improved clinical evidence
- Strengthening of post market surveillance (PMS)
- The power of a transparent EUDAMED database
So, how should companies prepare for the MDR, which internal processes need to be updated, which new documentation must be created, and what implementation activities should take priority? It helps to break the MDR preparation process into three phases:
- Activities that should start immediately, either due to the sequential nature of tasks, high priority or long lead times
- Activities that do not have to start immediately or require input from phase A, but must be completed by the time the MDR becomes enforceable
- Activities that are not strictly tied to the MDR enforcement date or can be implemented afterward
The table below shows key activities for each of the five topics mentioned above; a more in-depth discussion of each topic follows.
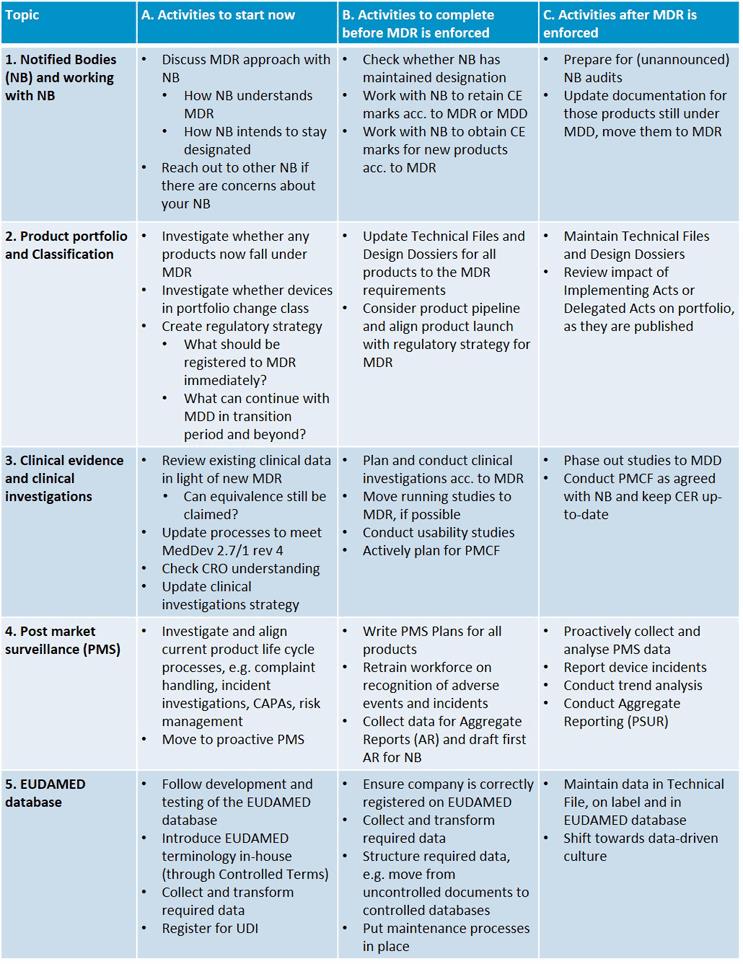
- Notified Body Interpretation/Preparation
The new MDR poses as many challenges to Notified Bodies as it does to manufacturers. Notified Bodies take on additional responsibilities, have to update their own procedures, require more subject matter expertise, and need to inform their customers of MDR-related changes. Additionally, Notified Bodies are under scrutiny and require re-designation to the MDR themselves. As such, the number of Notified Bodies has already fallen from more than 80 at their peak to the mid-50s now. The number is expected to fall further.
Notified Bodies are developing their own strategies to cope. For example, although manufacturers can ask to be certified to either the new MDR or the existing MDD during the transition period, Notified Bodies have indicated that it will be a challenge to run two parallel certification procedures. Thus, Notified Bodies may push for MDR certification earlier than is required by law. Furthermore, re-designation itself is an issue; it is not yet clear how long the process will take, or which Notified Bodies will be re-designated first. Some Notified Bodies are reducing the scope of their designation. Manufacturers risk that their Notified Body may not be designated to the MDR for a while, may no longer be accredited in the manufacturer’s product categories, or, at worst, may fail the re-designation process.
It is therefore important that manufacturers reach out to Notified Bodies early and enquire about approach and strategy. Notified Bodies already are inundated with calls; rumor has it that some have ceased to respond to requests as they set their strategies and explore implications. However, manufacturers need to keep pushing for surety that the Notified Body will continue to exist and that products remain in scope. Navigant already has noticed that Notified Bodies advocate different MDR strategies, in terms of approach, language, and pragmatism. Some bodies propose a practical approach, weighing the benefits of the MDR with the costs. Other Notified Bodies currently appear to interpret the MDR strictly, which would make placing certain low-risk products on the European market uneconomical.
In short, one of the first actions a manufacturer needs to undertake is to communicate with the Notified Body to discuss expectations and approaches. If no switch of Notified Body is required, close collaboration should ensure that a realistic approach is agreed upon and that products can be CE-marked in time. After May 2020, routine interaction with the Notified Body takes over, but regular interactions for additional data, post-market surveillance, new products, and inspections continue.
- MDR Implications On Product Portfolio
The MDR also applies to devices without an intended medical purpose, as listed in MDR Annex XVI. Additionally, classification rules, now in Annex VIII, have been extended. Thus, products previously considered borderline and/or outside the remit of the MDD will now be considered medical devices, or fall into a different risk class. For example, clinical decision support software — previously considered Class I — now will be in a higher risk Class, possibly even Class III, and, thus, subject to tougher documentation requirements. In general, the new set of classification rules is a step towards aligning US and EU classifications.
While it is possible to be certified to the MDD until the MDR enforcement day of May 26, 2020, and the MDD certification will allow products to remain in the European market for a further five years (void latest on May 27, 2024), any design changes after the enforcement day will force a switch from MDD to MDR. Manufacturers need to account for this in the product strategy. Manufacturers ought to remember that not all design changes are desired, plannable or foreseeable – for example, a product that is forced into a label change because of post-market surveillance data may require unexpected re-certification under MDR.
As a result, manufacturers need to assess the impact of the extended definitions, classification changes, and change expectations against their current and future product portfolios. A detailed analysis of new requirements is necessary. For example, a change in classification generally means additional documentation requirements, a more comprehensive risk analysis and, possibly, additional clinical work.
In terms of timing, the impact assessment on the portfolio should be carried out as soon as possible, as meeting any new requirements will take time. Any Technical Files or Design Dossiers need to be updated before certification to MDR. Future clinical work should be planned accordingly and updated processes must be implemented — for example, for the Quality Management System (according to EN ISO 13485:2016). After 2020, documentation has to be maintained to MDR standards; products still certified to MDD require switching to MDR.
- Need For Improved Clinical Evidence
Clinical evidence requirements in the MDR are significantly stricter. Equivalence to comparator devices must be scientifically justified for technical, biological, and clinical product characteristics. Additionally, the manufacturer needs to have access to the comparator’s data, including the Technical File and risk analysis. As competitors are unlikely to share this type of data, equivalency claims are likely to be successful only for products from the same organisation or partnership.
To acquire the required data, additional clinical investigations may be needed. Clinical investigation requirements have been integrated into the MDR and are also stricter than before. MedDev guideline 2.7.1 revision 4 for the MDD was published in 2016 [3] and gives a good indication of detail. Not all aspects of the MedDev guidance are relevant but, as a first activity, ensuring compliance with the guidance is a good step. To ensure alignment, manufacturers also should ensure that any partner Contract Research Organisations (CRO) fully understand the MDR. Most importantly, the portfolio should be investigated to where equivalency can still be claimed, and it should be determined where new clinical data is required.
Clinical investigations should be run to MDR requirements as soon as possible. Existing studies should consider which additional MDR burdens are worth implementing now, so the data can be used in future submissions. New studies should be set up with the MDR in mind, as clinical evaluation will become a continuous process.
- Strengthening Post-Market Surveillance
Guidelines on post-market surveillance (PMS) have been incorporated into the MDR. There are significant changes, not least of which is a reduction in the time required for notification of Competent Authorities of serious incidents — from 30 to 15 days. The regulation now includes PMS Plans for Class II and III devices, explicitly calling for post-market clinical follow-up (PMCF) and periodic safety update reports (PSUR). Class I manufacturers need to maintain PMS Reports.
These documentation requirements significantly increase the workload for manufacturers. Existing processes need to be streamlined to ensure reporting in 15 days. Complaint handling, often carried out by functions outside Regulatory Affairs, needs to be aligned with medical device reporting. In many cases, new processes for PSUR and PMCF need to be created. Additional resources likely are required.
Additionally, manufacturers ought to conduct an impact analysis of the new PMS requirements. Once the impact is known, procedures for creation of the required documents should be incorporated into the Quality Management System (QMS). Before the MDR’s enforcement date, these processes need to be in place and new PMS Plans, PMS Reports, and PMCF created for all products. After enforcement, proactive PMS and aggregate reporting via PSUR will be regular activities.
An interesting question is how the recipients (e.g., the Notified Bodies and competent authorities) will deal with the increased volume of PMS data and documentation. Clearly, for the system to be effective, it is not just the manufacturers who need to provide the required material; the recipients also must be able to receive and analyze the data. The effect of new PMS requirements on regulators, expert panels, and the overall safety of medical devices remains to be seen. Industry expects resource constraints here.
- A transparent EUDAMED database
The EUDAMED database has been operational for years. However, it is currently only accessible to competent authorities and Notified Bodies, and it does not contain the wide range of information required by the MDR. There have been plans to extend its capabilities for a while; the MDR seems to be the catalyst to make that happen, and further implementing acts on the EUDAMED database are expected. It is likely that information on all actors — including manufacturers, suppliers, authorised representatives, Notified Bodies, and competent authorities — eventually will be captured, as well as a large range of product information, from registration information to clinical investigation summaries and post-market safety data. In this regard, the EUDAMED database leads industry in the same direction as the European SPOR database for Identification of Medicinal Products (IDMP).
For manufacturers, the implications of an open, transparent European medical device database are significant. Lessons learned from the IDMP initiative so far include the introduction of a common terminology, standardized across an organisation and aligned to European terminology. Pharmaceutical manufacturers invest in their IT infrastructure to establish structured systems to collect and maintain the required data. New groups and positions for data management are being created.
Device manufacturers ought to consider how data (including “master data”) is currently stored across company systems, and how a common terminology across functions can be achieved. Manufacturers should plan for disparate data extraction, transformation, and loading into IT systems. Such activities require updated processes and a subtle shift in culture towards data-driven decision making. An advantage is that complying to new Unique Device Identification (UDI) requirements in Europe will become much easier. Of course, once a new system is in place, the registered data requires maintenance. Farsighted companies will see in the harmonization of data the opportunity to analyse, streamline, and automate processes and capabilities.
Manufacturers also should be aware that a transparent database gives competitors insights into products, and can be used by Notified Bodies to prepare for inspections. To avoid findings, products, Technical Files, and EUDAMED datasets need to become aligned and stay aligned. The pharma experience has shown that this is a difficult task.
Conclusion
The new European MDR has considerable impact on all actors in the medical device industry, including manufacturers and Notified Bodies. To some degree, manufacturers will just have to comply with the new rules. As this takes time and effort, preparations and impact assessments need to start now. Key items for early exploration are the future interaction with the Notified Body, impact on the product portfolio, collection of clinical evidence, updating of post-market surveillance procedures, and preparation for submission of transparent data to the EUDAMED database. Create a timeline for these activities; prudence suggests that the most urgent activities be started immediately.
About The Author
Tido Eger is an Associate Director in Navigant’s Healthcare and Life Sciences Disputes, Regulatory, Compliance and Investigations practice. He is an experienced healthcare consultant, focusing on medical device safety, regulatory compliance, vigilance, risk management, and organisational change management. He holds a PhD in Engineering from Cambridge University and a MEng in Engineering from Bristol University, and is a member of TOPRA.
References
[1] “Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EE,” Official Journal of the European Union, vol. 60, no. L117, pp. 1-175, 2017.
[2] “Council Directive 93/42/EEC of 14 June 1993 concerning Medical Devices,” Official Journal of the European Union, no. 169, pp. 1-60, 1993.
[3] “MedDev 2.7/1 rev.4 Clinical evaluation: a guide for manufacturers and notified bodies under directives 93/42/EEC and 90/385/EEC,” European Commission, 2016.