
The Design Controls + Risk Management Connection — Using Design Reviews Effectively
By Jon Speer, founder & VP of QA/RA, greenlight.guru

Design controls and risk management are key to the success of a medical device, in that they demonstrate that your product is safe and effective for its intended uses. Furthermore, there is a strong, complementary relationship between design controls and risk management, something we have discussed in this series’ previous installments.
Part one explored design control and risk management connections through intended use and user needs, while part two connected risk controls to design outputs, design verification, and design validation. In this installment, I will discuss best practices regarding design reviews, and how to incorporate risk management as a critical element helping to drive decisions.
Design Reviews — Breaking Down The Requirements
I like to start with the good ol’ design controls “waterfall” diagram to show how design reviews fit into the medical device product development paradigm:
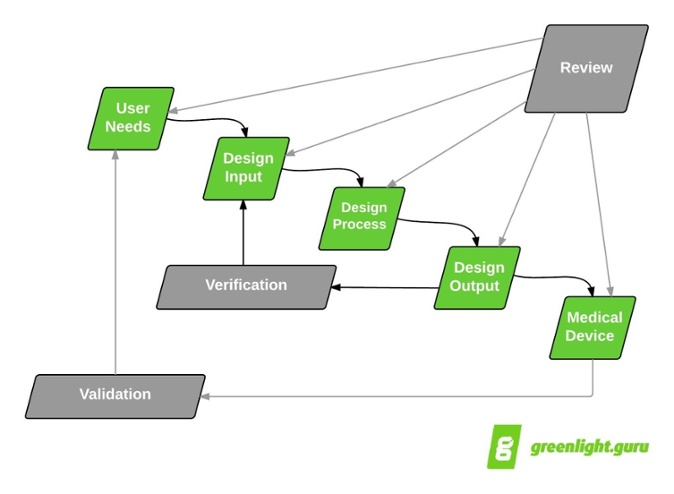
Design reviews are intended to encompass all aspects of design controls. Yes, that’s correct —all design control efforts should be included as part of a design review throughout the product development process. Consider what FDA 21 CFR part 820.30(e) and ISO 13485:2003 section 7.3.4 state about design reviews:
820.30(e) - Design review — Each manufacturer shall establish and maintain procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device's design development. The procedures shall ensure that participants at each design review include representatives of all functions concerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed. The results of a design review, including identification of the design, the date, and the individual(s) performing the review, shall be documented in the design history file (the DHF).
ISO 13485:2003 - section 7.3.4 - Design and development review — At suitable stages, systematic reviews of design and development shall be performed in accordance with planned arrangements (see 7.3.1)
a) to evaluate the ability of the results of design and development to meet requirements, and
b) to identify any problems and propose necessary actions.
Participants in such reviews shall include representatives of functions concerned with the design and development stage(s) being reviewed, as well as other specialist personnel (see 5.5.1 and 6.2.1).
Records of the results of the reviews and any necessary actions shall be maintained (see 4.2.4).
The key take-home points for design reviews are that they must:
- Be planned at suitable and appropriate stages during product development
- Include applicable functions for the stage being reviewed
- Include an “independent reviewer”
- Include documented records of the events
Design Review Vs Phase Review
Taken literally, the waterfall diagram suggests that design controls and subsequent design reviews follow a serial, or linear, progression.
Use of this methodology to manage product development projects is common in, but not unique to, the medical device industry. The “phase-gate model” (branded as Stage-Gate by Drs. Robert Cooper and Scott Edgett) bears a striking resemblance to the design control stages identified in the waterfall.
The basic premise is that each phase, or stage, is defined with minimum criteria and milestones. Before moving to the next phase, you need to satisfy that the criteria have been met, and the way to do so is via formal phase review. I bring this up for one primary reason. The medical device design controls process is more or less a phase gate approach, but with a slight twist.
The purpose of a phase review is to make a business decision — whether or not to continue funding a project, with dollars and resources, into the next phase of product development.
Design reviews serve a similar purpose, but dollars and resources are not the key metrics to monitor. With a medical device, the purpose at each stage is to demonstrate an acceptable level of safety and efficacy before continuing to the next stage. Design reviews are a mechanism used to assess and document these decisions.
It’s worth noting that there are ongoing debates on whether design reviews and phase reviews both are required for medical device product development. One point of view suggests that these should be separate events: Phase reviews analyze business decisions; Design reviews analyze design controls. The other point of view suggests that blending phase reviews with design reviews is perfectly fine.
Based on my 17+ years of experience with medical device product development, I recommend the latter approach. Do yourself a favor: Combine phase reviews and design reviews into a single meeting. Capture the design controls details on a design review form and other non-design control items in separate notes. We all have too many meetings as it is. Try to simplify things a bit when you can.
Must Medical Device Product Development Be Linear?
In a word: No.
Conventional wisdom suggests that linear product development processes are generally less risky (in a project sense of the word), versus processes that allow parallel activities. The en vogue product development practices involve lean and agile approaches, though, and you definitely can utilize such approaches in medical device product development.
But there are a few medical device design control absolutes that must occur and be documented:
- A design output must include or make reference to acceptance criteria.
- Design verification must demonstrate that design outputs meet design inputs. A design verification can only be conducted after design output / design input relationships have been established.
- Design validation must demonstrate that a product meets user needs. A design validation can only be conducted after product and user needs are defined.
- Design validation requires products to utilize production processes (or equivalents).
- All design controls must be part of design review(s).
Linking Risk Management To Design Reviews
Again, the primary purpose of both risk management and design controls is to ensure that your product is safe and effective for its intended uses. Demonstrating this requires objective evidence documented in a design history file and a risk management file.
Blending design controls and risk management, rather than treating them as entirely independent workflows, will improve your medical devices, and one way to bring these practices together is via design reviews.
As noted, you need to conduct design reviews at appropriate stages during design and development. I’ll let you decide on how often and how many. But since you are asking, my advice is to conduct a minimum of five design reviews, one at each of these stages: user needs, design inputs, design outputs, design verification, and design validation. Why? You need to show with objective evidence (i.e., documentation) that all design controls have been part of design reviews. The simplest and cleanest way to do so is by having separate design reviews for each of the major design control elements.
And, at each and every one of your design reviews, risk management should be the centerpiece of the event. Use risk management as a tool, rather than a checkbox activity. Use your ISO 14971-compliant approaches to drive risk-based decision-making as a practice within your medical device product development efforts. Identify where your product risks are, and discuss how to mitigate and control those risks via design controls; document these discussions as part of design reviews.
Understanding how to blend design controls and risk management efforts into a continuous stream will bring purpose and meaning to your product development efforts.
As regulatory bodies around the world harmonize their concepts of “risk-based approaches,” and tout the importance of complying with ISO 14971, you must evolve your own internal practices, as well.
About The Author
Jon D. Speer is the founder and VP of QA/RA at greenlight.guru, a software company that produces beautifully simple quality, compliance and risk management software exclusively for medical device companies. He is also the founder of Creo Quality, a consultancy that specializes in assisting startup medical device companies with product development, quality systems, regulatory compliance, and project management. Jon started his career in the medical device industry over 16 years ago as a product development engineer after receiving his BS in chemical engineering from Rose-Hulman Institute of Technology.