
The Design Controls + Risk Management Connection — Verification, Validation, & Risk Controls
By Jon Speer, founder & VP of QA/RA, greenlight.guru

The previous article in this series addressed design control and risk management connections through intended use and user needs — specifically, how these items are key to identifying hazards, hazardous situations, and foreseeable sequence of events.
To recap, intended use leads to user needs, which lead to design inputs. Think of design inputs as a “contract” by which medical devices are designed and developed. Intended use also helps define the scope of a design & development plan, as well as the scope of a risk management plan.
Let me also remind you of the high-level risk management process overview defined in ISO 14971:
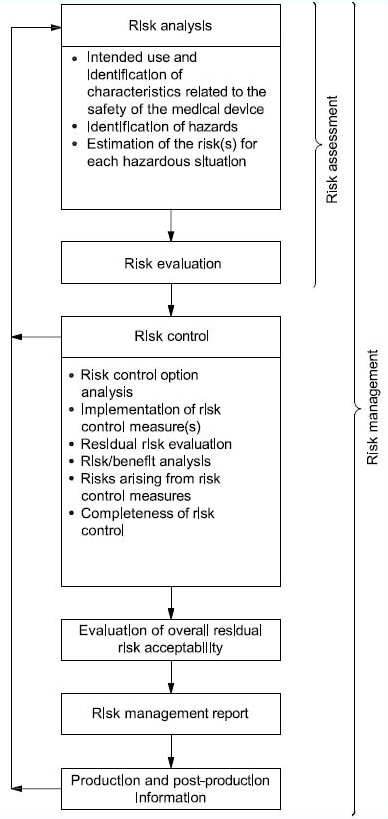
Here, I’ll continue the journey of demonstrating how design controls and risk management should flow seamlessly back and forth to improve safety and reduce risks associated with medical devices, tying in the connections that design inputs, design outputs, design verification, and design validation have with risk controls.
From the perspective of risk management, you have estimated the risks of each hazardous situation by determining severity of potential harm and its probability of occurrence.
You established risk acceptability criteria earlier, during the risk management planning phase of product development. Chances are, you have some type of risk acceptability matrix or chart where you have defined regions of both acceptable and unacceptable risks.
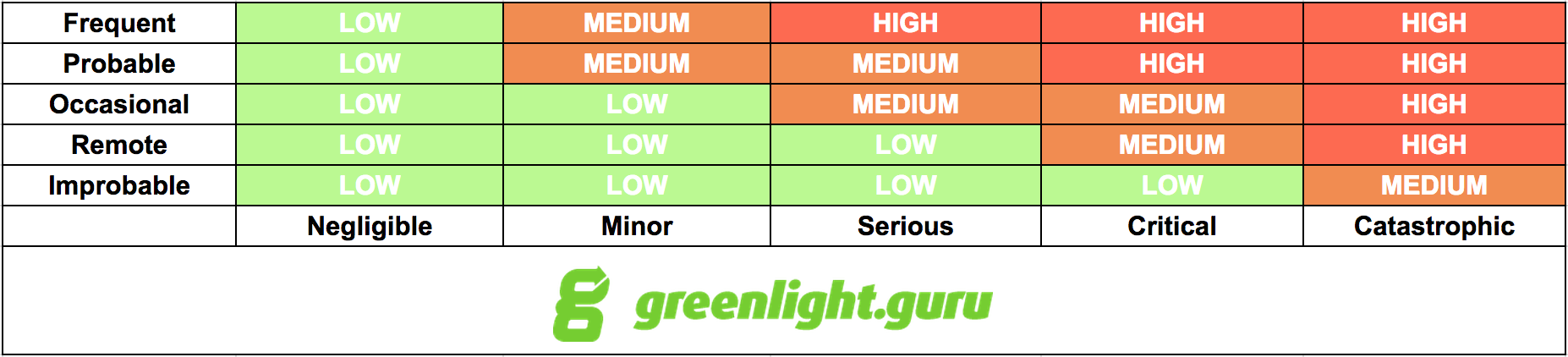
Based on the risk acceptability criteria you have established, there is one significant question you need to answer at this stage: Is risk reduction necessary? If you have made the design controls and risk management connection, then the answer to this question will be a driving force for the next several stages of your medical device product development.
ALARP vs ALAP
Depending on where in the world you are selling medical devices, the decisions driving risk reduction can take on different meanings.
Specifically, for Europe, EN ISO 14971:2012 indicates that all risks must be reduced “as low as possible” (ALAP). If you are following ISO 14971:2007 (basically, everywhere except EU), then all unacceptable risks should be reduced to acceptable status, or to “as low as reasonably practicable” (ALARP). By the same token, risks initially in the ALARP “zone” should ideally be reduced, as well.
The Risk Control “Phase”
When you have identified hazardous situations requiring risk reduction, you enter the risk control phase of the risk management process. According to ISO 14971, risk control is the process in which decisions are made and measures implemented by which risks are reduced to, or maintained within, specified levels.
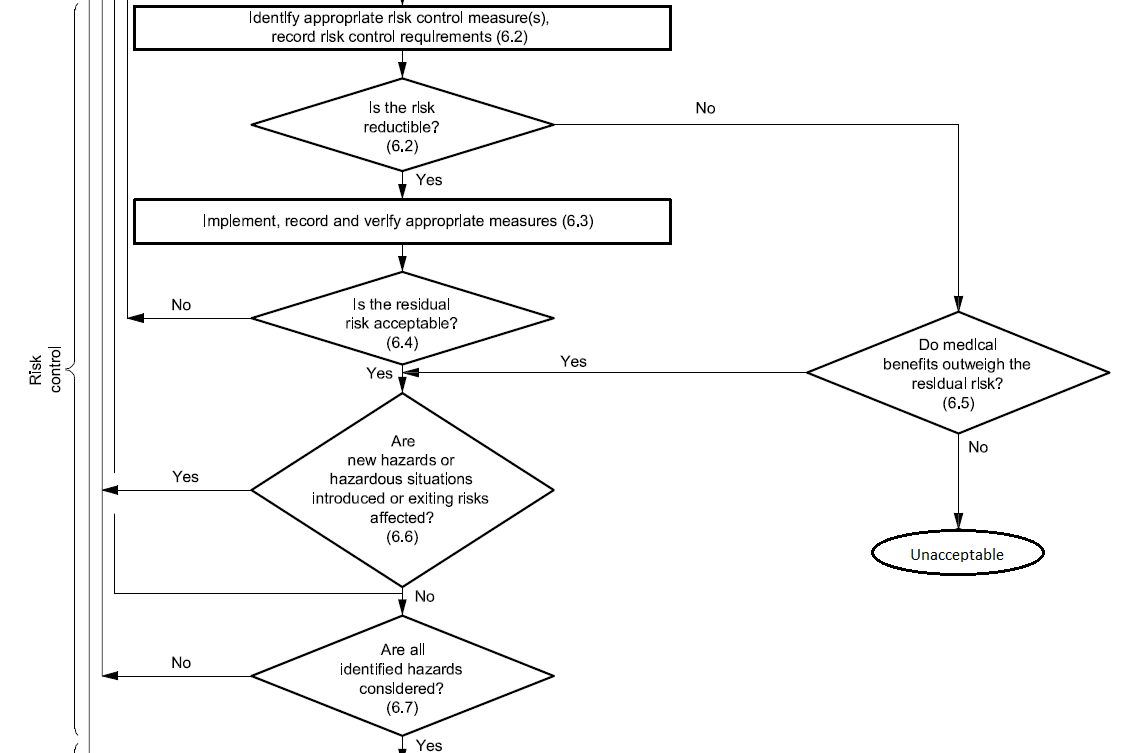
Risk Control — Considering Your Options
In order to reduce risks to acceptable levels, you need to identify possible options — risk control measures — that are appropriate and substantial. There are three basic types, or levels, of risk controls you should consider, listed here in order of priority:
- Inherent safety by design — Ensure that the design of the medical device reduces and/or eliminates the probability of harmful occurrences altogether.
- Protective measures in the medical device and/or manufacturing process – Examples of this could include redundant features, safety mechanisms, etc., with the intent to reduce the occurrence of harm.
- Information for safety – This category of risk controls — which includes labeling, instructions for use, training materials, and the like — is largely regarded as least effective, so much so that EN ISO 14971:2012 does not allow you to use this as a means for risk control because information for safety is a general requirement of medical devices.
Risk Controls And The Design Controls Connection
As you can see, there is a strong connection the levels of risk control measures and design controls.
When you identify risk control measures, these items can drive changes and revisions to your design inputs. Perhaps your design inputs can be better clarified and stated. Or, perhaps you need to include additional design inputs.
Design outputs, too, can be significantly influenced by your risk control measures. Remember that the design outputs established during medical device product development serve as the preliminary device master record (DMR) for the product. In other words, the design outputs are the “recipe” for your medical device. Risk control measures relating to design outputs could lead to adding new safety features into your product and providing more clarity and definition in design output documents.
KEY TIP: I like to do a first pass through risk analysis, risk evaluation, and risk controls after I have my user needs and design inputs defined, and BEFORE doing too much work on my design outputs, design verification, and design validation. Why? I want to know what my initial risk levels are before creating overly complicated drawings and specifications, and before conducting expensive — and maybe unnecessary — verification and validation (V&V) testing.
Identifying risk control measures should help you define what type of design verification activities will be necessary to demonstrate that your design outputs meet your design inputs, and to prove your medical device is safe. Design verification activities are also instrumental in providing some objective evidence to support probability of events that could lead to harm.
As with design verification, risk control measures can also help shape what type of design validation will be necessary to prove the product addresses user needs. Design validation can serve as a means to determine if hazards and hazardous situations are likely during intended use. Results of design verification and design validation will be influential when estimating and evaluating residual risks.
Residual Risk Acceptability
After identifying all risk control measures and implementing them, the next step is to evaluate and estimate the residual risks: Did you successfully reduce risks to acceptable levels? If not, consider additional risk controls.
Or, maybe you can conduct a risk/benefit analysis where you evaluate and weigh the medical benefits your device provides against the risks. If you choose this path, document the explanation. Also, note that risk/benefit analysis is another area where EN ISO 14971:2012 differs. If you have devices in EU, then you need to document a risk/benefit analysis for all of your risks.
Did your risk controls introduce new hazards and hazardous situations? If so, analyze, evaluate, and estimate the risks from these new scenarios.
Conclusions
Risk control measures are the key to identifying ways to mitigate and reduce your product’s risks to acceptable levels. Risk controls provide a means to help you develop your medical device “recipe” through design outputs, to prove these outputs meet design inputs via design verifications, and to prove your medical device meets end user needs.
As I have stated before, medical devices are intended to save and improve quality of life. Following sound design controls and risk management processes helps ensure that the devices you design, develop, manufacture, and sell are as safe and effective as possible.
About The Author
Jon D. Speer is the founder and VP of QA/RA at greenlight.guru, a software company that produces beautifully simple quality, compliance and risk management software exclusively for medical device companies. He is also the founder of Creo Quality, a consultancy that specializes in assisting startup medical device companies with product development, quality systems, regulatory compliance, and project management. Jon started his career in the medical device industry over 16 years ago as a product development engineer after receiving his BS in chemical engineering from Rose-Hulman Institute of Technology.