
The Intersection Of ISO 13485 And ISO 14971 Under The Proposed FDA QMSR
By Edwin Bills and Christie Johnson

The FDA proposed on Feb. 22, 2022, an update to the Quality System Regulation1 that was released in 1996. The proposal is to reference ISO 13485:2016 Medical devices — Quality management systems — Requirements for regulatory purposes,2 which is in its third edition since its 1996 release. The FDA QSR has not been updated since its 1996 release and needs to be brought to the current state of the art. Incidentally, the first edition of ISO 13485 was released at about the same time as the initial QSR, so 13485 has been updated twice while the FDA’s regulation has not been updated.
Both the FDA’s 1996 QSR and ISO’s 13485:1996 documents were originally based on ISO 9001:1994,3 which was organized in the same manner as the current 21 CFR 820 with 20 clauses of requirements. ISO 13485 strayed away from that structure in the 2016 edition after the 2008 edition of ISO 9001 changed the structure to the process model with eight clauses. The ISO medical device quality system technical committee chose not to update to the 2015 edition of ISO 9001 as it had strayed too far from the regulatory purpose of ISO 13485. They did update the 2016 edition along the lines of the regulatory purpose.
It is interesting to note that the FDA has participated heavily in the development and revision of ISO 13485, yet it was not able to update its own regulation. Also, the FDA participated in the MDSAP audit program4 and accepted audits of the manufacturer’s quality system to ISO 13485:2016 in place of routine FDA QSR inspections. The agency, with its deep knowledge of the ISO standard, apparently is now comfortable using it in place of 21 CFR 820, the Quality System Regulation.
One of the key statements in the FDA’s proposal, on page 10, is its intention to implement ISO 13485 as a key part of the new Quality Management System Regulation (QMSR): “FDA has long recognized the value of, and has been exploring ways to effect, global harmonization for the regulation of devices. For example, the FDA has actively participated in the development of internationally harmonized documents and standards on risk management since their inception, including the development of the Global Harmonization Task Force (GHTF) guidance document, ‘Implementation of Risk Management Principles and Activities Within a Quality Management System’5, dated May 20, 2005, which outlines the integration of a risk management system into a QMS (Ref. 6). FDA also participated in the development of the various versions of ISO 14971 ‘Medical Devices--Application of Risk Management to Medical Devices.’”
Importantly, the FDA said, in a presentation at MedCon 2023 in Columbus, Ohio, on April 26, 2023, that its rationale for switching to incorporating by reference ISO 13485:2016 into the new QMSR included:
- Modernized QMS principles.
- Greater integration of risk management activities
- Globally harmonized requirements
In this article, we propose to show the intersections of ISO 13485:2016 medical devices quality systems standard and ISO 14971:2019 medical device risk management standard, which the FDA implied as key to the new regulation in the MedCon statement.
Aligning On The Definition of Risk
Looking at the original 21 CFR 820, the FDA only used the word “risk” once as part of the term “risk analysis.” For those who have read the 38-page preamble to the QSR (the regulation is 14 pages), you would know that the FDA expected far more of the manufacturer in the area of risk than the one item shown in the regulation. The preamble contained 200 categories of responses to the 10,000 comments received on the 1994 proposal to adopt the QSR. (In contrast, the agency only received 68 responses to the proposed QMSR, and many of those were concerned with the transition period, perhaps indicating more acceptance of the QMSR.) In those preamble comments there are 36 uses of the word “risk,” including 12 to explain the FDA’s position on the use of “risk analysis” in the regulation, two in Purchasing, one in Servicing, and three in CAPA. Two uses appear where the FDA discounts the use of “economic risk” in the analysis of the medical device and one in regard to risk of design-related failures. Other references use the word “risk” as a modifier, such as “low-risk devices”. In contrast, ISO 13485 uses the word “risk” 18 times in the requirements of the standard, as opposed to the FDA’s single use in the current 21 CFR 820.
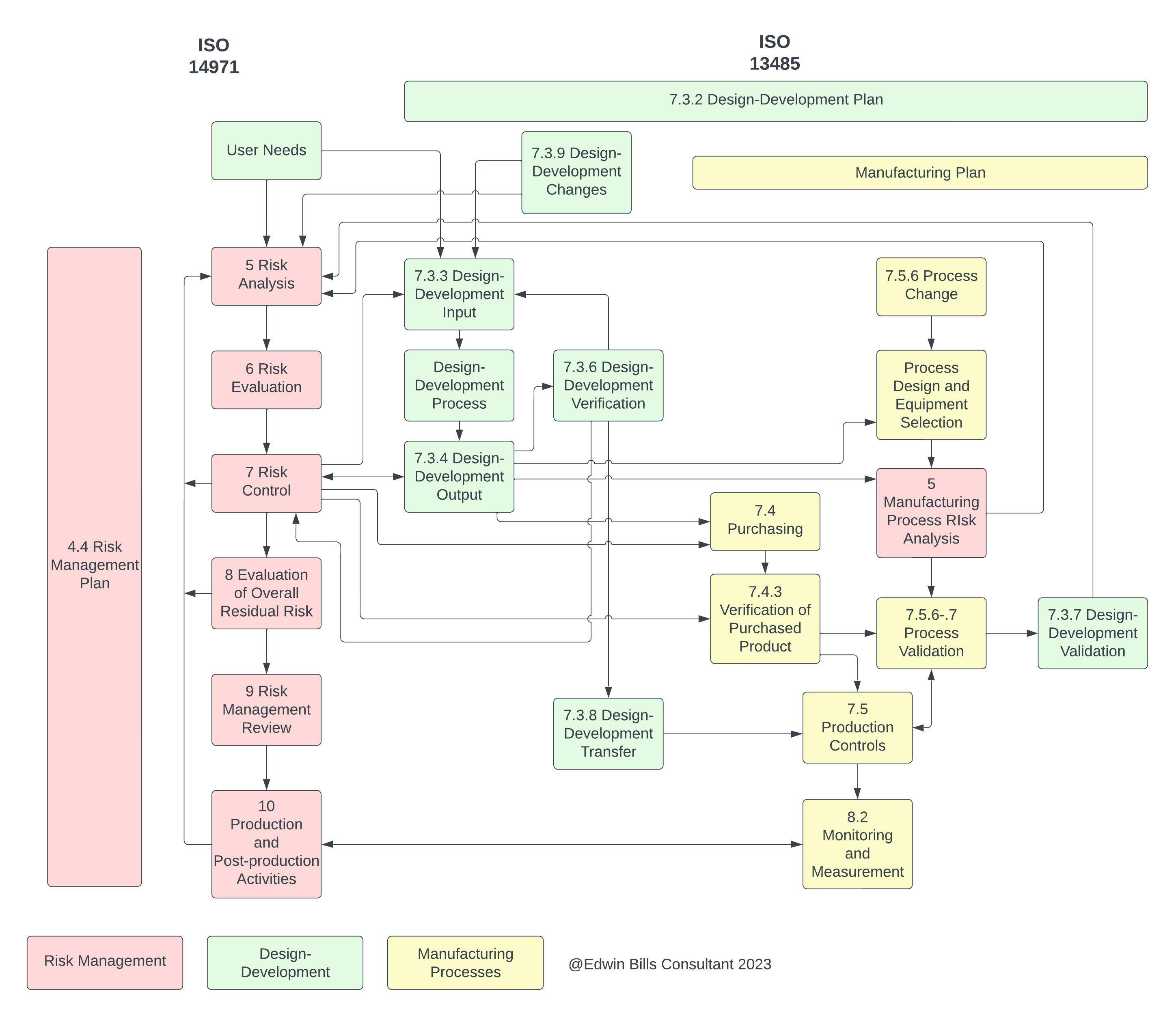
Figure 1: A partial diagram of the intersections of ISO 14971 and ISO 13485. Click on image to enlarge.
If we examine ISO 13485 and its use of “risk,” we can see how the two ISO standards intersect. Figure 1 was created to show where the active parts of the two standards intersect. That is, each of the two has parts that “set the stage,” so to speak, but the parts shown above in Figure 1, are where activities take place. It would be difficult to show the “set the stage” parts in this diagram, and we have elected to show only those intersections where an activity such as design input takes place. These active parts are those that would be unique to a specific medical device created during the product realization process, where the “set the stage” items would be a common set that applies to all devices going through the product realization process.
The “set the stage” requirements include such items as personnel, documentation, and management, for instance, and are not shown on the diagram. This shows how complex the relationship between ISO 13485 and ISO 14971 is, with information moving back and forth between the two at different times as a new product is being developed and a current on-market product is being maintained. It is important to note that risk management activities must occur before Design-Development Input to meet the requirements of ISO 13485:2016 7.3.3 c) on providing the outputs of risk management as design inputs.
ISO 13485:2016 in Clause 3.16 refers to the definition of “risk” in ISO 14971, though it calls out the 2007 edition, of course, since that was the edition in effect at the time 13485:2016 was created. The FDA’s 21 CFR 820 did not provide a definition of the term “risk” in the regulation or the preamble, but it did refer to the ISO committee, which had just begun work on developing a risk management standard, ISO TC 210 WG4, later combined with IEC to form ISO TC 210 JWG1, which continues today as the committee assigned to maintain ISO 14971 and the risk management guidance ISO TR 24971. While the current FDA regulation does not define “risk,” we can see in its proposed move to ISO 13485 in the QMSR, it will be using the ISO 14971 “risk” definition that is part of ISO 13485 definitions, and since the 2007 and 2019 definitions are the same, that will not be an issue. Similar thinking applies to the only other definition from ISO 14971:2007, “risk management.”
Where Does ISO 13485 Reference ISO 14971?
Now that we are aligned on the definition of risk, we need to understand where ISO 13485 calls out ISO 14971. It is not a “normative requirement,” which would mean compliance with ISO 14971 was required to meet the requirements of ISO 13485. The definition in 3.16 of 13485 does, in Note 1, indicate that “This definition of ‘risk’ differs from the definition given in ISO 9000:2015” which is an enterprise risk management definition from ISO 31000, and is not suitable for product safety risk management. However, ISO 13485 makes the following distinction in Clause 0.2 Clarification of concepts, “Where the term ‘risk’ is used the application of the term within the scope of this international standard pertains to the safety or performance requirements of the medical device or meeting applicable regulatory requirements.”
The first place “risk” is encountered in ISO 13485 requirements, after definitions, is in Clause 4 Quality management system 4.1.2 b) states, “The organization shall apply a risk-based approach to the control of the appropriate processes needed for the quality management system.” “Risk-based approach” is undefined in the ISO 13485 standard. However, the document ISO 13485:2016 Medical Devices – A Practical Guide (Advice from ISO/TC 210)6 provides several pages of discussion on the topic, including “the use of risk is in the context of the safety and performance of the medical device and meeting regulatory requirements and not financial risks or risks to business performance.” The next encounter with “risk” is in 4.1.5, where the discussion is on control of outsourced processes where the controls are “proportionate to the risk involved.” The next risk discussion is in 4.1.6 on software validation and revalidation of computer software in the quality management system, where, again, the term “proportionate to the risk” is used to determine the level of activities needed. We next go to Clause 6.2 Human resources, where the discussion on personnel being competent in a Note states, “The methodology used to check effectiveness is proportionate to the risk associated to the work for which training or other action is being provided.”
Product Realization
From there we go to Clause 7 Product realization, which is where the actual product realization activities begin. Here we find in the second paragraph of 7.1 Planning of product realization: “The organization shall document one or more processes for risk management in product realization. Records of risk management activities shall be maintained.” At the end of 7.1 is a Note, which is probably the most direct reference we can find in ISO 13485: “Further information can be found in ISO 14971.”
From here we go on to 7.3.3 Design and development inputs, where c) states applicable outputs of risk management are considered to be requirements inputs to design-development. This would mean that risk management activities, from risk analysis through risk evaluation up to risk control, must be completed prior to design input to provide the safety requirements for design input. That is something new for many people, especially those who use failure modes and effects analysis (FMEA), a reliability tool, as their only risk analysis tool, as FMEA cannot be done until there is design output information available.
Since risk management is considered to be an iterative activity throughout the product lifecycle, the design requirements may be updated as new information is learned during the product realization process, though it becomes more expensive in both time and money later in the process to make product and process changes to update a design from this new information. Additionally, it is important that all risk analyses from any tool used to identify hazards and hazardous situations be entered into the single Risk Management File for the product (Figure 2). This file must be kept available and up to date, including all risk information from all sources and including all parts of the medical device throughout the entire product lifecycle, until there are no more devices of this type in the field. The file must be traceable from the hazard, to the risk analysis, to the risk evaluation, to the verification of the implementation of risk controls, and the results of the residual risk evaluation (ISO 14971:2019 4.5)
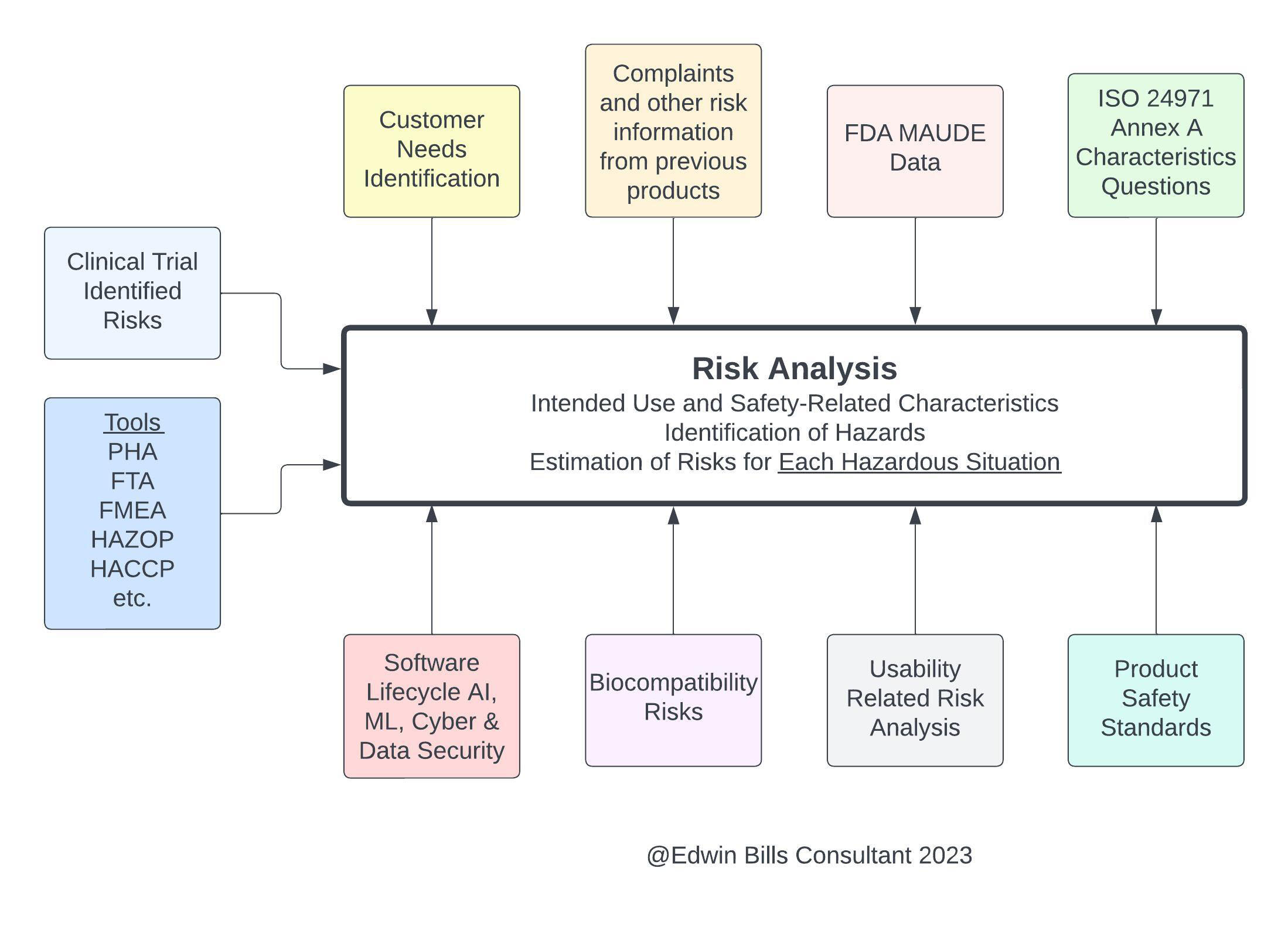
Figure 2: Inputs to ISO 14971:2019 include all risk analyses from any tool. Click on image to enlarge.
Production and Post-production
Now let’s leave Design-Development, for which most companies have implemented risk management close to what is required in both ISO 14971 and ISO 13485, and venture into other areas of risk management. If you look at Clause 8 of ISO 13485, which is Measurement, analysis and improvement, you will find references in 8.2.1 Feedback for feeding back information into risk management from production and postproduction. The purpose of this feedback is to maintain product requirements as well as product realization or improvement processes. In a number of places of the document, we find discussions of maintaining safety, which of course is the inverse of risk (risk goes down as safety goes up), so anywhere we encounter “safety” we must consider risk management methods to maintain or improve that level of safety. This is also where the concept of life cycle risk management is introduced in 13485, which of course is also discussed in Clause 10 of ISO 14971:2019 Production and post-production activities. The two standards both developed their respective Clause 8 and Clause 10 activities from the GHTF SG3:N18 document on Corrective and Preventive Action, so they are pretty much aligned here.
Although outside the scope of this article, the ISO TR 20416:20207 technical report on post-market-surveillance shows that alignment in the process of setting requirements and providing deliverables in its Figure 1. The purpose of post-market surveillance is stated in Clause 4 of ISO TR 20416 as Monitoring safety and performance, Meeting regulatory requirements, and Contributing to life cycle management. ISO TR 20416 is where the two standards (14971 and 13485) requirements to address regulatory requirements for post-market surveillance are identified and are too expansive to be discussed in this article.
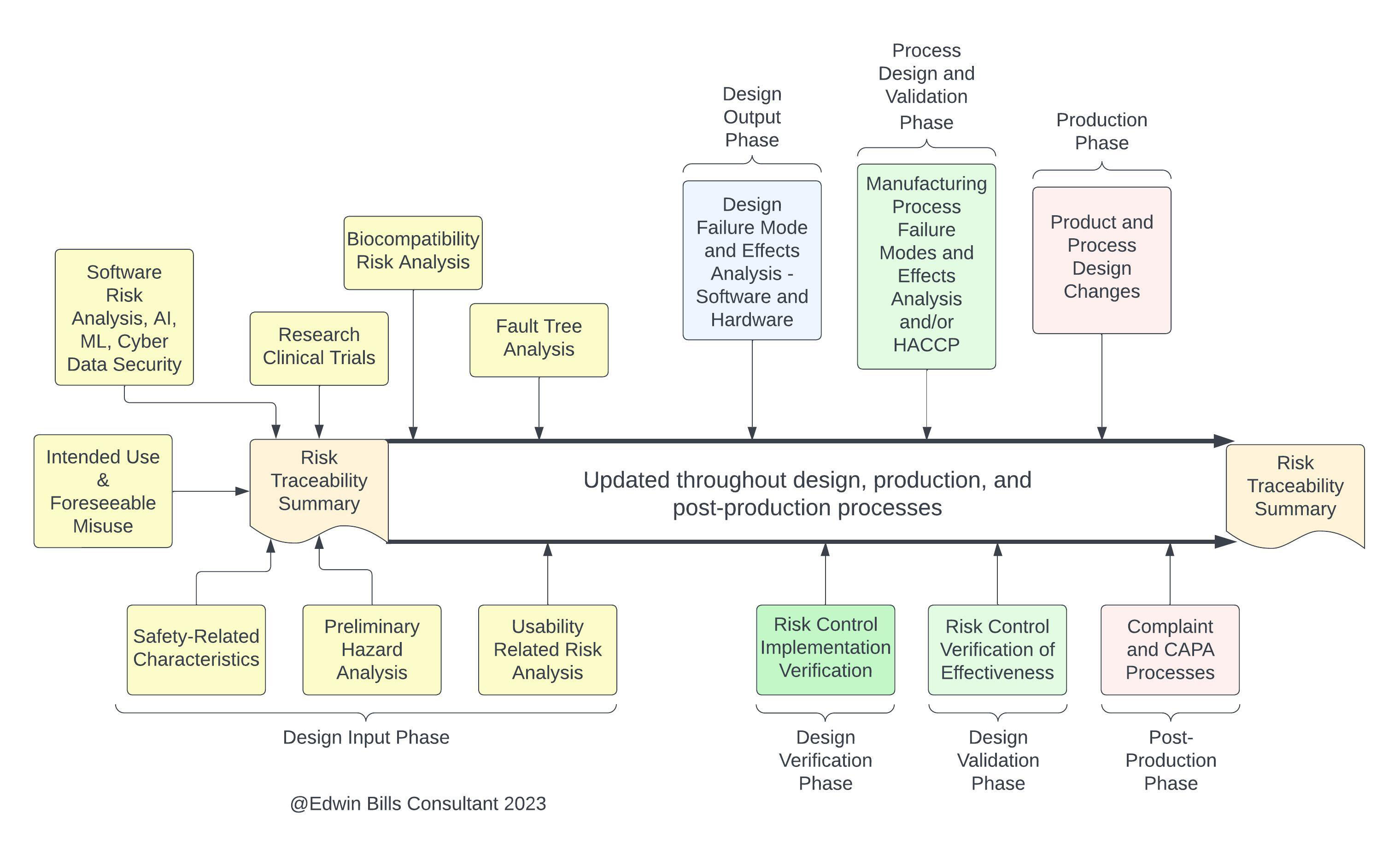
Figure 3: Risk activities at different product realization stages. Click on image to enlarge.
Conclusion
Both ISO 14971:2019 and ISO 13485:2016 were developed with product safety in mind, as well as how the two standards would work together to reach that goal. As both standards define complex processes, as we can see in Figure 1 of this article, it takes a great deal of effort to create a quality system that includes a risk management system and results in safe and effective products over the entire life cycle of medical devices, including IVDs and, especially, any devices that include software.
The development of an effective system requires support and involvement of top management to provide resources including a team of knowledgeable individuals with varying backgrounds. These personnel would include such diverse areas as design, risk, clinical, medical, manufacturing, regulatory, legal, and post-market support to create a risk management system that produces safe medical devices. One group cannot do this job without the participation of everyone who touches product safety.
A risk management system should produce products that are lower cost and in a more timely manner than products produced in a system where risk documents are simply put in the file at the end of the design phase, which we often find today. That approach – placing documents in the file after the product is designed and sent to manufacturing “to get it out there” – eventually results in product redesigns, both during design transfer and in post-production, recalls, and product liability costs, which together are much higher than under a risk management system.
With the upcoming revision of 21 CFR 820 into the QMSR, it is important that manufacturers conduct gap assessments for both ISO 13485:2016 and ISO 14971:2019 requirements to assure they will be ready when implementation of the new QMSR occurs. Following the gap assessments, a review of the proposed regulation as it appears in the Federal Register will be appropriate to note the additional items the FDA proposes to add in the QMSR and any other changes, such as in definitions. Implementation of the new regulation should reduce the burden on manufacturers from having to comply with the differing current regulations and should simplify documentation requirements as well.
References
- Quality System Regulation 21 CFR 820:1996, US FDA
- 2. ISO 13485:2016 Medical devices-Quality management systems-Requirements for regulatory purposes, ISO 2016, Geneva, Switzerland
- ISO 9001:1994 Quality management systems-Requirements, ISO 1994, Geneva, Switzerland
- www.fda.gov/medical-devices/cdrh-international-programs/single-audit-program-mdsap
- Implementation of Risk Management Principles and Activities Within a Quality Management System, Global Harmonization Task Force, 2005
- ISO 13485:2016 Medical devices Advice from ISO/TC 210, ISO 2017, Geneva, Switzerland
- ISO TR 20416:2020-Medical devices-Post-market surveillance for manufacturers
 About The Authors:
About The Authors:
Edwin L. Bills, RAC, ASQ Fellow, CQE, CQA, CMQ/OE, has been a member of ISO TC 210 JWG1 for more than 20 years. This is the ISO group responsible for medical device risk management and the creation and maintenance of ISO 14971:2019, the risk management standard for medical devices, and ISO TR 24971:2020, the accompanying risk management guidance.
 Christie Johnson is a partner at Prodct LLC and director of quality at myBiometry. She is an advisor in product risk management and quality management systems to early-stage medtech startups and serves as a content expert to NIH's RADx initiative for the advancement of COVID-19 diagnostics. She was recently accepted into the ISO TC 210 committee for advancement of risk management standards for medical devices, ISO 14971:2019 and ISO/TR 24971:2020.
Christie Johnson is a partner at Prodct LLC and director of quality at myBiometry. She is an advisor in product risk management and quality management systems to early-stage medtech startups and serves as a content expert to NIH's RADx initiative for the advancement of COVID-19 diagnostics. She was recently accepted into the ISO TC 210 committee for advancement of risk management standards for medical devices, ISO 14971:2019 and ISO/TR 24971:2020.