
Understanding Australia's Regulatory Framework For SaMD
By Raj Tumbad, Freyr Solutions

Regulatory bodies around the world are continuously refining their regulatory frameworks to ensure the safety and efficacy of software-based medical devices and software as a medical device (SaMD). This article examines the important changes and subsequent impacts of software medical device regulations in Australia.
The oversight of medical devices in Australia has traditionally been under the governance of the Therapeutic Goods Administration (TGA). On Feb. 25, 2021, the TGA implemented reforms in the regulation of software-based medical devices, and certain software-based medical devices were carved out (through either an exemption or exclusion) from the scope of the TGA regulation. These reforms covered a wide range of software, including SaMD, which functions as a medical device on its own. The goal was to clarify the scope of regulated software products, introduce new classification rules, and update the essential principles governing software-based medical devices.
SaMD Regulation: Main Changes In Legislation And Classification Impacts
One significant change has been the transition to a risk-based classification system. These regulations are being changed to differentiate software medical devices based on their potential risks to patients and users. This shift acknowledges the varying complexities of software, ensuring that low-risk software products do not undergo unnecessary regulatory burdens, while high-risk ones receive thorough scrutiny. The classification of software-based medical devices that provide a diagnosis or screen for a disease or condition is determined by whether the device directly delivers the diagnostic/screening or assists the health professional in making such determinations based on its information. For software-based medical devices intended to provide information to monitor the state or progression of a disease or condition, the classification depends on both the potential risk to public health and whether the information could indicate if a person is in “danger.”1
Another significant change is carving out certain software-based medical devices by exempting or excluding them from the regulation. Some clinical decision support systems (CDSSs) have been exempted. Exempted software is considered a medical device but is not subject to all the regulatory requirements. For example, software designed for tasks such as comparing a specific patient's symptoms and test results with existing clinical practice guidelines, or surgical workflow software that outlines the procedural steps of a surgery to a surgeon. In contrast, excluded software is not classified as a medical device and is therefore exempted from all the TGA regulatory requirements. For example, a web-based application that offers reference information on specific diseases or conditions according to a health practitioner's input of their patient's symptoms. The application does not provide an indication of probability, red flags, or priorities.2
More Rigorous Clinical Claims About Safety And Performance
The foremost concern in software-based devices is patient safety. Any SaMD that fails to deliver on its intended purpose or introduces erroneous data can lead to misdiagnosis or inappropriate treatment, jeopardizing patients’ well-being. Rigorous clinical claims are essential to ensuring that SaMD meets the highest standards of safety. The TGA expects a high level of scrutiny applied to the serious conditions or claims that are commensurate with the level of risk associated with the intended use/intended purpose/claims/indications for the SaMD, for instance, a SaMD that leads to death/severe deterioration/poses a moderate risk to public health. The TGA considers the applications based on the response provided/mentioned in the manufacturer’s technical documentation, like information for use (IFU), device labels, and device advertising materials/brochures, and seeks internal clinical advice in relation to the claims and intended use of the device. The Australian classification rules for SaMD will consider the user of the device as well as the severity of the risk associated with the use of the product and the severity of the disease/condition the device is intended to be used for.
TGA Assessment Of SaMD: Post Submission Queries
If there are any concerns about the SaMD or information provided in the application, the TGA may raise any queries regarding the device. In some instances, the application will be selected for audit. For some applications, an audit is mandatory under the legislation. Others may be selected for auditing at the discretion of the delegate or as a free audit. The sponsor will be notified if an audit occurs and asked to provide further information and supporting documentation and. In the case of a mandatory audit, the sponsor will receive an invoice for the assessment fee.
Table 1: Types of the TGA Audits and their Respective Objectives3
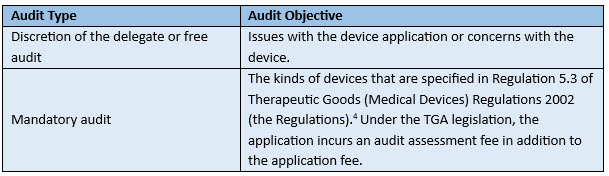
Delegate or Free Audit for Medical Device: The TGA requests the applicable documents, such as clarification on the Australian classification rule selected by the manufacturer, pictorial images of the device, IFU, brochures, advertising materials, and device labels with the Australian sponsor details.
Mandatory Audit: There are two levels of audit assessment (level 1 and level 2). Based on the nature of the device application, the TGA will determine which level applies. The TGA requests documents like the manufacturer’s declaration of conformity (DoC), device details, clinical evidence, and risk management report. The TGA has target time frames of 30 to 60 business days for medical device application audits. Also, under the Therapeutic Goods (Medical Devices) Regulations 2002, specific device applications are subject to mandatory auditing unless the manufacturer has a TGA conformity assessment certificate, the EU MDR 2017/745 certification, or an inclusion in the Australian Register of Therapeutic Goods (ARTG) as only an export medical device.
Australia has adopted a classification system that aligns closely with the European Union's Medical Device Regulation (EU MDR). However, recent changes in software regulations have introduced nuances that can impact the classification of SaMD. In most circumstances, the classification will be the same as in the EU, except in cases where the risk to public health is greater than the risk to an individual. Despite the fact that the device is MDR CE certified, the TGA assesses it with a higher level of regulatory scrutiny. Depending on the intended purpose/claims/indications of the software, the devices may be classified as high-risk in Australia, i.e., the device is Class IIa MDR CE certified, but under new software standards it may be categorized as Class IIb in Australia. For instance, if a manufacturer possesses an MDR Certified Class IIa software designed to detect fractures in osteoarticular X-rays of specific organs such as the hand, shoulder, leg, ribs, and spine. However, in compliance with new software regulations in Australia, the TGA classifies both rib and spinal fractures as a "disease or condition that may lead to death/severe deterioration without urgent treatment." Consequently, the SaMD is categorized as Class IIb, particularly considering urgent scenarios, such as superior rib fractures (first or second rib, which can be associated with mediastinal injury, including to the aorta), multiple (three or more) rib fractures, rib fractures associated with symptoms of internal injury (e.g., difficulty breathing, hematuria), and rib fractures in frail or elderly patients. All of these situations would necessitate a referral to the emergency department. Therefore, the TGA classifies it as Class IIb in Australia. The TGA considers the applications based on the response provided/mentioned in the manufacturer’s technical documentation and seeks internal clinical advice in relation to the claims and intended use of the device.
TGA Rejection Challenges And How To Avoid Them
Manufacturers and sponsors must conduct a thorough risk assessment if the SaMD has been impacted by the new classification rules in Australia and examine if the SaMD is up-classified. Sponsors must ensure that they provide appropriate conformity assessment certificates or supporting evidence (from comparable overseas regulators/assessment bodies for medical devices) to pass the TGA’s preliminary assessment during the ARTG listing.
Conclusion
The implementation of software requirements by the TGA has significantly heightened the scrutiny level for device approval and corresponding TGA audits. Even leading global SaMD manufacturers are encountering challenges with device registrations under these new software regulations. Ultimately, the path to achieving successful SaMD TGA approval hinges on seeking guidance from experts and gaining a thorough understanding of the new SaMD requirements. Sponsors and manufacturers must be agile and responsive to these changes, ensuring that their SaMDs meet the software regulatory requirements. By reassessing their SaMD in light of new changes, they can navigate the regulatory landscape effectively and contribute to the safe and effective use of medical software in Australia's healthcare system.
References
- Regulatory changes for software based medical devices (Version 1.2, August 2021)
- Clinical decision support software - Scope and examples (general guidance Version 1.1, October 2021)
- Auditing of medical devices, including IVD medical device applications (July 12, 2023)
- Therapeutic Goods (Medical Devices) Regulations 2002
About The Author:

Raj Tumbad leads medical device approval projects at Freyr Solutions and is a domain expert in the field of Australia's medical device regulations. He has extensive experience executing quality and regulatory strategies for medical device and in vitro diagnostics (IVD) companies, including European Union Medical Device Regulation (EU MDR), quality management systems (QMS), and post-market surveillance (PMS) of medical devices, including software and AI-based medical devices.