
Understanding The New MHRA Requirements For Medical Devices In Great Britain
By Marcelo Trevino, independent expert

Recent developments at U.K.’s Medicines & Healthcare products Regulatory Agency (MHRA) as a result of EU MDR extensions and other factors have caused the agency to update its transition arrangement dates for CE marked devices. In this article, I will summarize the most recent changes and provide guidance to medical device manufacturers on steps to follow in order to commercialize medical device products in the U.K. under the new regulatory framework.
On Jan. 9, 2024, the MHRA revealed a road map for medical device regulation, aiming to prioritize patient safety and establish a new framework by 2025. The new framework includes a focus on implantable devices, artificial intelligence, cybersecurity, and early disease detection diagnostics. By introducing new regulations, the agency aims to enhance international harmonization while keeping up with technological advancements.
Several months ago, the U.K. government announced an extension of CE marking recognition acceptance for several businesses; however, it also was made clear that this recognition does not apply to medical devices or in vitro diagnostic devices. MHRA has emphasized that transition arrangements for CE marked medical devices are still applicable and has provided a road map that includes clarification of conformity assessments and economic operator requirements.
The graphic below provided by the agency provides a visual representation of the new 2024–2025 road map:
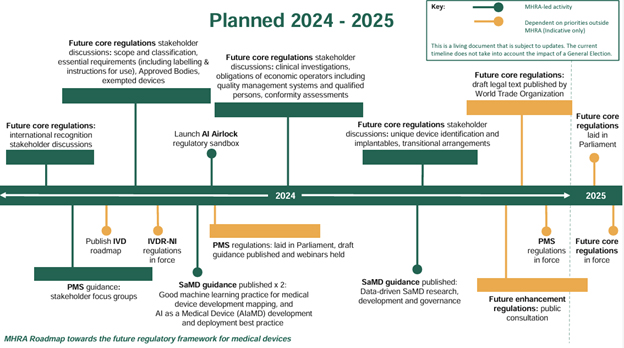
Figure found in the MHRA’s road map toward the future regulatory framework for medical devices.
MHRA postponed enforcement of its legislation until 2025. For now, there is a standstill period that has been extended until June 30, 2025. The most recent EU MDR 2023/607 extension also has been accepted. As a result, EU MDR and EU IVDR certificates will be accepted until June 30, 2030, with certain provisions.
The government has put in place the Medical Devices (Amendment) (Great Britain) Regulations 2023 legislation, which amends the Medical Device Regulations 2002 (SI 2002 No 618, as amended) (U.K. MDR) to extend the acceptance of CE marked medical devices on the Great Britain market. Transitional arrangements were published in April 2023 and implementation began in May 2023. These requirements will apply to devices in the GB market through EU directives, the EU Regulation, or the U.K. MDR 2002.
The amendments seek to introduce clearer, risk-proportionate post-market surveillance (PMS) requirements. This will help to improve the ability of both the manufacturer and the MHRA to identify issues with medical devices placed in the Great Britain market and, where necessary, take appropriate action to safeguard public health.
Some of the key changes include:
a. An increase in the scope of devices that must comply with the new PMS requirements; this includes CE marked devices.
b. Details on what must be included as part of a PMS system, including the methods for collecting PMS data to support improved capturing of PMS data and harmonization across manufacturers.
c. Enhanced serious incident reporting obligations for manufacturers to support the detection of safety issues sooner.
d. More stringent requirements for manufacturers to conduct periodic reviews of their PMS data, including for implantable medical devices. This aims to support manufacturers in earlier detection of trends/signals that may have an impact on the safety of a medical device.
e. Improved coordination and collaboration between manufacturers, U.K. Approved Bodies, and the MHRA to support regulatory oversight.
This MHRA link provides a graphic with the timelines for placement of CE marked medical devices on the Great Britain market under the Medical Devices (Amendment) (Great Britain) Regulations 2023.
Paths To Place A Device On The GB Market
Currently, there are two paths to place a device on the GB market:
- Path 1: Conformity assessment as per EU legislation (directives or regulations) plus EU Declaration of Conformance (DoC), CE mark, and registration with MHRA; or
- Path 2: Conformity assessment per U.K. Conformity Assessment (UKCA) legislation plus UKCA mark and registration with MHRA.
While Great Britain encompasses England, Scotland, and Wales, Northern Ireland, which is part of the U.K., still recognizes and will continue recognizing EU CE marking under the regulations and directives.
If an organization is using directives, a conformity assessment according to EU/MDD/AIMDD/IVDD is required. Class Is, Class Im, Class IIa, and Class IIb require the following:
- uploading Article 120 extension confirmation with MHRA
- U.K. responsible person (UKRP) appointed if required
- conditions under 2023/607 shall be met
- compliance with PMS requirements (a draft statutory instrument that will be enforced in mid-2024)
Statutory instruments (SIs) are a form of legislation that allow the provisions of an Act of Parliament to be subsequently brought into force or altered without Parliament having to pass a new Act. They are also referred to as secondary, delegated, or subordinate legislation.
The following products can be placed on the market until June 30, 2028, if they rely on the EU MDD: Class IIb, implantable well-established technology, IIa, Is, Im, and up-classified Class I devices that were covered under EU MDD DoC. After June 30, 2028, these products will have to comply with the Medical Device Amendments Regulations (2023) and the new statutory instrument that will be officially published later this year.
Class IIb implantable (non-well-established technology or non-WET), Class III, and active implantable medical devices (AIMD) have the same requirements but can only be placed on the market under the EU MDD scheme until Dec. 31, 2027.
If an organization is using EU Medical Device Regulations (EU MDR) to place a device on the GB market, the following applies:
- For all medical device classifications, a UKRP shall be appointed if required and the PMS requirements as per the draft SI. Medical device products that meet EU MDR/IVDR can be placed on the market until June 30, 2030, if they have a valid CE Marking. After June 30, 2030, these products will have to comply with the Medical Device Amendments Regulations (2023) and the new statutory instrument that will be officially published later this year.
- In vitro diagnostic devices under List A, List B, and Self Tests require an appointed UKRP and compliance with PMS requirements (draft SI). IVDD Class D devices can be placed on the market until May 26, 2025, if they have a valid CE Marking. After May 26, 2025, these products will have to comply with the Medical Device Amendments Regulations (2023) and the new statutory instrument that will be officially published later this year.
The rest of IVD products can be placed on the market until May 26, 2030, or until their certificate expires, whichever is sooner. After May 26, 2030, these products will have to comply with the Medical Device Amendments Regulations (2023) and the new statutory instrument that will be officially published later this year.
Medical device manufacturers can access guidance from the MHRA here. As a first step, manufacturers must register with MHRA and upload an Article 120 extension confirmation.
Labeling Requirements
A U.K. conformity assessment mark (UKCA) mark is required to be placed on medical devices conforming with UKCA requirements. Placement of the label can be on the device, the sales packaging for the device where practical and appropriate, and on the instructions for use for the device. Article 17 of the EU directives can continue to be applied to comply with UKCA.
Devices that are placed on the U.K. market based on EU certification or CE marking cannot use a UKCA mark and need to maintain their CE mark label. Manufacturers outside the U.K. must appoint a UKRP, but it is not mandatory to identify the UKRP on the labeling or IFU.
Devices that are placed on the U.K. market based on UKCA certification must use the UKCA mark on the device or its sterile pack where practical and appropriate, except if they are custom made devices. UKCA marking must be affixed to any sales packaging for the device and the instructions for use for the device. Manufacturers located outside the U.K. must also appoint a UKRP and identify the UKRP on their labeling or in the IFU. If a U.K. accredited body is used as part of the conformity assessment, it must also be included in the label next to the UKCA mark.
Details of the U.K. responsible person should be provided on any devices bearing the UKCA mark in accordance with the requirements laid out in the regulations. For AIMDs, this would be on the sales packaging and for IVDs it could be either on the label, outer packaging, or on the IFU.
Post Market Surveillance Statutory Instrument (PMS SI)
The Post Market Surveillance Statutory Instrument (PMS SI) aims to improve patient safety while creating better harmonization across the industry, increasing scrutiny and regulatory oversight, enhancing reporting obligations for manufacturers, imposing more stringent PMS requirements depending on the classification of the device, and improving coordination between manufacturers, Approved Bodies, and the Secretary of State.
Currently, under the Medical Devices Regulations 2002 (SI 2002 No 618, as amended) once a medical device has been placed on the market, the manufacturer must continually monitor the performance of the medical device. However, there are limited regulatory details in the legislation on the steps that manufacturers are required to follow to comply with PMS and vigilance obligations. Details are provided in a guidance and not in the legislation, which has created inconsistencies in the way manufacturers conduct their PMS activities.
Contents of the draft include requirements to implement the following:
- PMS system
- PMS plan
- Preventive and corrective actions
- Initial reporting of serious incidents
- Investigation and final reporting of serious incidents
- Field safety corrective actions (FSCA) and field safety notice (FSN)
- Field safety corrective actions (FSCA) outside GB
- PMS report (self-declared)
- PSURs (all other classes)
- Trend reporting
- Reports received by Secretary of State
- Analysis of information
- Retention of PMS documentation
The government intends to publish a final version of the PMS SI in June 2024, and it is expected that will become applicable toward the end of 2024. A guidance also will be published once the final version is released.
Manufacturer Responsibilities Under The New PMS SI
- Ensuring that the organization’s QMS covers all aspects of the statutory instruments, and that QMS assessments also cover implementation of the statutory instruments.
- Producing periodic safety update reports (PSUR) for each device placed on the market or put into service. Single PSURs can be generated for a group of devices if they have the same intended purpose and technology.
- Ensuring that the PSUR includes U.K. specific requirements and that it is provided to the Secretary of State within three days upon request.
- Updating PSURs every year for all device classes, except IIa devices, which should be updated every two years.
Future Legislation In The U.K. For Medical Devices
The U.K. government recently responded to a consultation on the future regulation of medical devices where it confirmed that the MHRA will be acting soon to implement the following:
- Introduce several improvements for implantable medical devices, including up-classifying them, which will result in more stringent pre- and post-market requirements and requiring manufacturers to provide implant cards to enable patients to know which device they have had implanted.
- Ensuring that devices have a unique device identifier (UDI).
- Changing the classification of several types of devices, specifically, increasing the class of certain software as a medical device and aligning IVD classifications with those of the International Medical Device Regulators Forum (IMDRF).
- Introducing a framework for international recognition, enabling swifter access for devices already approved by comparable regulators as well as for those who have Medical Device Single Audit Program (MDSAP) certificates.
Conclusion
MHRA’s recently announced regulatory framework road map aims to create a comprehensive plan for significant improvements to the regulatory framework. The agency will continue to focus on high-risk and innovative devices, particularly software and devices that include artificial intelligence technology.
While transition timelines provide more time for manufacturers to comply with the new requirements, they rely on manufacturers meeting EU 2023/607 requirements, and all PMS requirements from the SI requirements still need to be met. Additionally, if manufacturers are using the directive route to comply, they cannot introduce significant changes to their device. Alignment with EU MDR/IVDR can facilitate an easier implementation.
Manufacturers can have an opportunity to combine assessments if they already hold other certifications like MDSAP or EU MDR/IVDR. This could be a great advantage if the Approved Certification Body is also the EU Notified Body and MDSAP provider.
A final version of the new PMS SI will be published in a few months, along with a guidance document on implementation for manufacturers. The agency continues working with industry and various medical device associations to support an effective implementation of the new regulations.
About The Author:
 Marcelo Trevino has more than 25 years of experience in global regulatory affairs, quality, and compliance, serving in senior leadership roles while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, medical imaging/surgical navigation, in vitro diagnostic devices, and medical device sterilization and disinfection products. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR/IVDR, MDSAP). He holds a BS in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. Trevino is also a certified Medical Device Master Auditor and Master Auditor in Quality Management Systems by Exemplar Global. He has experience working on Lean Six Sigma Projects and many quality/regulatory affairs initiatives in the U.S. and around the world, including third-party auditing through Notified Bodies, supplier audits, risk management, process validation, and remediation. He can be reached at marcelotrevino@outlook.com or on LinkedIn.
Marcelo Trevino has more than 25 years of experience in global regulatory affairs, quality, and compliance, serving in senior leadership roles while managing a variety of medical devices: surgical heart valves, patient monitoring devices, insulin pump therapies, surgical instruments, orthopedics, medical imaging/surgical navigation, in vitro diagnostic devices, and medical device sterilization and disinfection products. He has an extensive knowledge of medical device management systems and medical device regulations worldwide (ISO 13485:2016, ISO 14971:2019, EU MDR/IVDR, MDSAP). He holds a BS in industrial and systems engineering and an MBA in supply chain management from the W.P. Carey School of Business at Arizona State University. Trevino is also a certified Medical Device Master Auditor and Master Auditor in Quality Management Systems by Exemplar Global. He has experience working on Lean Six Sigma Projects and many quality/regulatory affairs initiatives in the U.S. and around the world, including third-party auditing through Notified Bodies, supplier audits, risk management, process validation, and remediation. He can be reached at marcelotrevino@outlook.com or on LinkedIn.