
Updating Your Quality Management System (QMS) For UDI
By Dan O'Leary, President, Ombu Enterprises

The Unique Device Identification (UDI) rule touches just about all parts of the FDA regulations for medical device makers. These are called conforming amendments, because they make the other parts conform to the concepts of the UDI rule. For example, complaints (820.198) and medical device reports (MDRs) (Part 803) need to include the UDI.
In addition to the explicit requirements, there are also important implicit requirements. For example, many companies use a database to record complaint information. The explicit rule change requires the addition of database fields for UDI. The implicit requirement is to revalidate the software application following 820.70(i). In addition, there will be changes to procedures for complaint handling, which will require additional training.
The date to put UDI on devices, labels, and load GUDID is the compliance date, which varies by the device class and other characteristics (e.g., implantable vs. reused, and processed between uses).
The date to change procedures is the effective date, and it was December 23, 2013. In other words, you should have updated your procedures, databases, etc. However, the guidance document Unique Device Identifier System: Frequently Asked Questions, Vol. 1 [PDF] explains the practical effect. Section A.13 says, “For example, the amendments to parts 820 [Quality System Regulation] and 822 [Postmarket Surveillance] will have no practical effect until 1 year after publication of the final rule, when class III devices become subject to UDI labeling requirements.”
FDA has not issued a policy on enforcement discretion, so, presumably, an investigator could ask for your UDI procedures before the compliance date for your device.
Procedures, Document Control, And Personnel
Device manufacturers develop procedures and work instructions to ensure correct and uniform application of the quality system requirements. This follows from the FDA’s view as stated in the preamble to the Quality System Regulation (QSR), which states, “FDA also notes that the quality system regulation is premised on the theory that adequate written procedures, which are implemented appropriately, will likely ensure the safety and effectiveness of the device.” While specific to Part 820, this principle carries throughout all the parts of the regulations. In each case, identify your implementing procedures and update them for UDI.
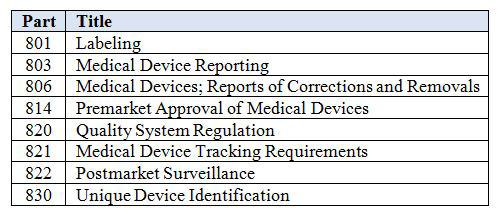
Table 1 lists the parts of the FDA regulations affected by the UDI rule. Some may not apply to every device manufacturer. For example, Part 821 on medical device tracking applies to a limited number of companies. On the other hand, Part 806 on corrections and removals applies to all device manufacturers.
Table 1: Parts Affected by the UDI Rule
As you develop new procedures and modify existing ones for UDI, you need to consider three important aspects: review and approval, training, and distribution. These apply to any change; they are not unique to UDI. However, UDI implementation provides a good opportunity to ensure your systems are effective.
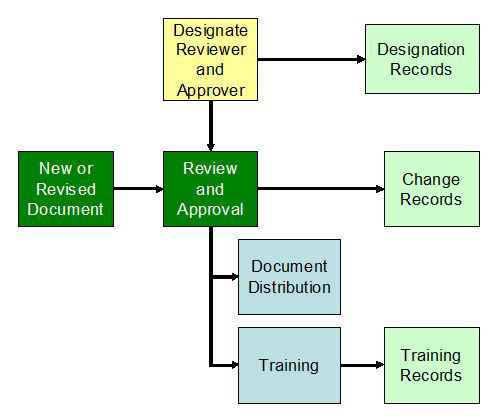
Figure 1: New or revised document
A designated individual needs to review and approve the documents. While the approver is often the process owner, a reviewer should be knowledgeable about UDI. You must develop an organizational structure to maintain FDA’s database, the GUDID. It would be reasonable for the regulatory contact or a GUDID coordinator to review all new and changed documents that implement UDI.
Because UDI changes many aspects of current processes, such as labeling inspection in 820.120(b), be sure to train the people in these processes and create training records following your documented procedures.
Competency And Training
While UDI will add elements to some existing processes, GUDID creates new processes and new roles. As a result, there will be new procedures, definitions of competency, designated individuals, and training records. A manufacturer must establish a regulatory contact, at least one GUDID coordinator, and at least one GUDID labeler data entry (LDE) user. During the process to set up an account, the manufacturer will identify the people by name and provide contact information to FDA’s Center for Devices and Radiological Health (CDRH).
For each of these new roles, define the necessary education, background, training, and experience following 820.25(a). Subsequently, identify training needs, train the people in these roles, and document the training records.
Another area of concern is new skills required by designated individuals. A prime example is the person who performs the 820.120(b) labeling inspection. That person must examine the device label for accuracy, including the UDI. Since the UDI is in easily readable plain text and automatic data identification and capture (AIDC), the designated individual must be able to read both. If, for example, the AIDC uses a bar code, then the designated individual needs to scan the bar code and verify the information it contains. This requires an update to the definition of competency, changes to procedures, training, and training records. In addition, this designated individual is performing a verification activity, so he or she needs to be aware of potential defects and errors.
Databases And Software Validation
In some cases, a manufacturer might use a database or computer system to record information. Two common applications are complaints and service records. Both require the UDI as part of the record.
The UDI includes both the device identifier (DI) and as many as five production identifiers (PIs). A good strategy would expand the database to include all six elements, even if you don’t currently expect to use all of them. If conditions change, you would not need to do an additional expansion and validation.
Because this is a change to the database, 820.70(i) states, “All software changes shall be validated before approval and issuance. These validation activities and results shall be documented.”
Quality Audits
Part of your UDI implementation plan should include quality audits shortly after implementation. After making the changes, conduct an audit of each area to ensure effective implementation. Schedule the audit two or three months after the changes go into effect to give the processes time to absorb them. The audit should include documentation revisions, distribution to points of use, correct records generated by the process operator, and complete training records.
In addition, schedule a set of product audits after the compliance dates, to verify the label and GUDID implementation.
Part 820
This part, the Quality System Regulation, has a number of explicit changes, such as labeling inspection, complaint records, and servicing records.
There are also some implicit requirements, such as new or revised procedures, training records, and software validation.
One major implicit requirement is design changes under 820.30(i). The DI represents the version or model of the device. If a change modifies the device outside established limits, then the manufacturer assigns a new DI and creates a new DI record in GUDID. Design change procedures should include a checklist to determine if the change creates a new version or model, and it should leave a record of the decision-making process.
Part 803
MDRs require a UDI on the Form 3500A. This means the manufacturer must update the procedures and train the people involved in the process.
Part 806
Reports of a correction or removal require a UDI. Some corrections or removals are not reportable, but the record of why it is not reportable includes the UDI.
Other Parts
As shown in Table 1, UDI affects many parts of the device regulations. Determine which parts affect your quality system, download the updated regulations, and search for the term “UDI”. This will help you identify the explicit changes. Identify your procedures and sections that the UDI rule could affect. Be sure to consider any implicit changes based on your methods and processes.