
Will We Have Harmonized Standards By The MDR's Date Of Application?
By Leonard Eisner, Eisner Safety Consultants

The EU Commission’s draft standardization request to EU Standardization Organizations was issued June 26, 2019 — 11 months prior to the MDR’s May 26, 2020, date of application. Specifically, the EU Commission requested that CEN (EU Committee for Standardization, the EU equivalent to ISO) and CENELEC (EU Committee for Electrotechnical Standardization, the EU equivalent to IEC) draft revisions to their existing standards, adapt some international standards, and issue new standards in support of MDR (2017/745) & IVDR (2017/746).
The request document rationalizes that “Voluntary harmonised standards should help to ensure high level of protection of health for patients and users throughout the Union and thus contribute to the free movement of devices in the Union. Given that such standards are technology-neutral and performance-based, they also contribute to ensuring equal conditions of competition among economic operators dealing with devices…”
This article focuses primarily on the MDR but will touch on IVDR. As such, not all applicable IVDR references will be cited. Consider this article a gap assessment on MDR proposed harmonized standards. From its second page onward, the request document includes draft text for the Commission Implementing Decision (pages 2 – 25). This article deals with text and tables from this draft text.
The draft text directly references the General Safety & Performance Requirements in Annex I, Chapter I, point 1 of both the MDR & IVDR, stating “Technical specifications included in the standards should support the attainment of these objectives (Annex I, Chapter I, point 1).” While there is no direct reference to the EU Harmonized Standards Annex Zs in the request document, the request embraces their intent in stating “each harmonised standard…should…specify the extent to which the technical specifications included in the standard aim to cover one or several requirements set out in” the MDR or IVDR.
This notification is based on the requirements noted in the MDR & IVDR on Harmonized Standards (Article 8(1) of the MDR & IVDR); it references the EU Standardizations Regulation 1025/2012 and the Commission Staff Working Document…work program for European standardization for 2018, as well as several other documents. The 2018 EU work program states three reasons for new standards applicable to the MDR & IVDR: new device types, new regulated practices, and more detailed safety and performance requirements.
The EU Commission has requested that CEN and CENELEC provide feedback on the request document by July 25, 2019. However, the request is not a complete adoption of the existing MDD, AIMDD, and IVDD list of harmonized standards.
The draft text specifies, rather, that “this Decision should cover horizontal standards addressing the needs of the widest scope of…economic operators and to allow a subsequent alignment of semi-horizontal and device-specific standards which may derive from or complement the horizontal standards.” [underlined and italicized text is used here for emphasis; it does not appear in the original text.]
Only five standards in the request document are slated for completion by the MDR’s date of application — not very functional for implementation of the MDR with regard to use of Harmonized Standards. All remaining requested standards for the MDR have a May 27, 2024 date of application (four years after the MDR date of application).
The draft text indicates “The references to standards listed in Table 1 of Annex I [of the request document]…correspond with the most recent versions of those standards published by CEN and CENELEC on the date of this Decision.” As noted in the table below, not all standards listed in the Proposed column are the most current. Annex I, Table 2 is discussed later in this article.
The table below highlights some of the standards listed in Annex I, Table 1. While some of the titles are omitted in the table below, more detail is provided than in Annex I, Table 1 of the request document; ideally, this will help readers to understand:
- the equivalent international standard (IS)
- whether draft standards are in development per the applicable ESO and/or International Standards Organization
- the current MDD / AIMDD Harmonized Standard
- additional relevant considerations
Some of the proposed standards are not what most would consider ‘state of the art’ (a non-defined term, but one that is referenced multiple times in the MDD, AIMDD, IVDD, MDR & IVDR), as they are not based on the most current published IEC, ISO, CEN, or CENELEC standard. This leads to the question, which version of the standard should a manufacturer use when assessing their devices?
As the MDR’s Annex I, item 1 (General Safety & Performance Requirements) requires ‘state of the art,’ manufacturers must follow the most current version of a standard, even if it has not been harmonized under the MDR. I have recommended this approach to my clients for years under the MDD/AIMDD. I also have heard stories that you can get a different answer, depending on your EU Notified Body. However, I don’t see any text in the MDR that deals directly with this issue — other than the ‘state of the art’ requirement in Annex I, item 1, which some (but not all) Notified Bodies require at this time.
For some proposed standards on the list, the CEN or CENELEC databases (db) note that the standard is expected to be cited in the Official Journal of the European Union (OJEU) to the MDD, AIMDD, or IVDD. However, this is unlikely to occur, as resources are now focused on MDR & IVDR implementation. This means the proposed standard or the version noted hasn’t been cited in the OJEU previously. Still, I will not focus on the past (MDD, AIMDD, or IVDD), since many Notified Bodies authorized for the MDD, AIMDD, &/or IVDD have stopped accepting any further work for these Directives. This article is focused on the MDR’s future.
Click table to enlarge
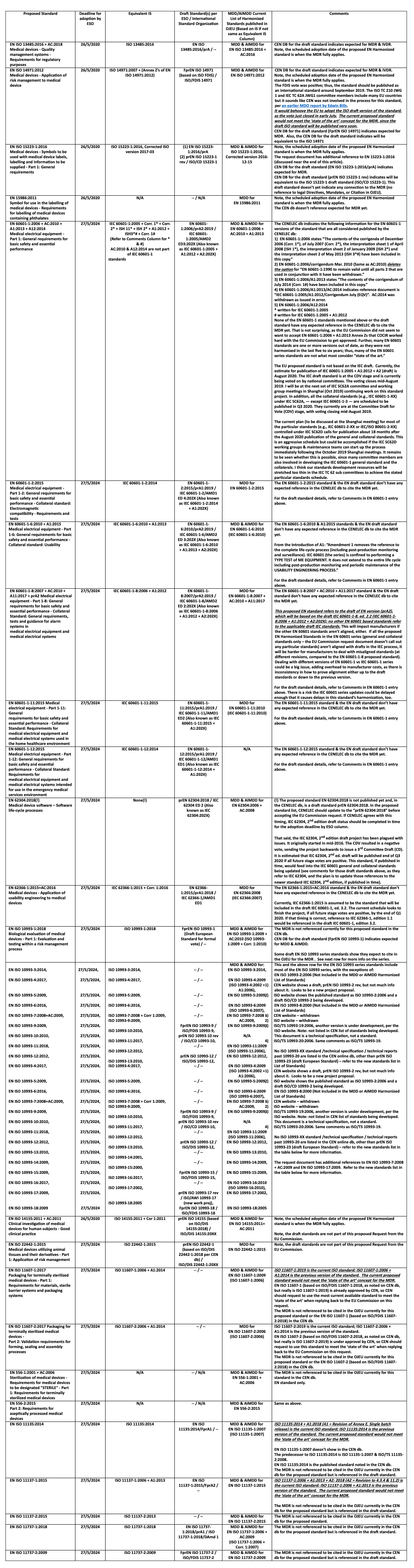
The EU Commission request document’s Annex I, Table 2 lists new standards to be drafted in support of the MDR, as well as the European Standards Organizations’ (ESOs) proposed dates of adoption:
Click table to enlarge:

The EU Commission’s request document — in Annex III, Part B — asks for specific additional updates or drafts relevant to four standards listed in Annexes I & II:
- EN ISO 15223-1:2016 “Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements” — Add symbols indicating whether a device is a medical device or an in-vitro diagnostic medical device to facilitate application of Section 23.2(q) of Chapter III in Annex I to Regulation (EU) 2017/745 or Section 20.2(e) of Chapter III in Annex I to Regulation (EU) 2017/746, as appropriate.
- EN ISO 10993-7:2008 + AC:2009 “Biological evaluation of medical devices - Part 7: Ethylene oxide sterilization residuals” and EN ISO 10993-17:2009 “Part 17: Establishment of allowable limits for leachable substance” — The update request asks that “the method of calculation of residue limits for EtO sterilant” of these applicable standards “be modified in such a way as to take into account” the weight of the patient, especially considering neonates and other patients.
- A request that “CEN shall draft a standard describing technical solutions for safety-engineered mechanisms to be applied in design and manufacture of devices” that “ensure compliance with points 11.1 and 22.2” of Annex I Chapter II of the MDR — “The standard shall apply to devices which intended use is the administration and/or extraction of body/blood fluids and/or medicinal substances.” ISO 23908 is referenced in Annex III, Part B and is listed in Annex 1, Table 2 (new standards list for MDR) in the request document. EN ISO 23908:2013 is in the CEN db. EN ISO 23908:2013 was never harmonized under the MDD.
I am hopeful that more EU Harmonized Standards will be proposed and actually developed by the date of application – five is not enough — but I must admit to being pessimistic based on the MDR implementation process not being timely and effective. Specifically, consider the limited number of Notified Bodies (NBs) designated with less than 11 months remaining the date of application — only two designated after 1½ years, one of which is in the UK (and the BREXIT drama may negate that designated NB by November) — and the numerous NBs dropping out of the designation process recently.
Note, too, the standards harmonization process’ slow pace over the last six years, as well as the lack of implementing acts and guidance documents released. Cloudier still is the status of EUDAMED development and the eventual impact of BREXIT (a significant number of all medical devices in the EU use a UK-based NB).
I hope I am wrong in my pessimism, as it would be horrible for a well-intentioned regulation to limit medical device accessibility in the EU. Per the EU Commission Factsheet for healthcare professional and health institutions: “These changes could have consequences for the availability of medical devices for health institutions. For instance, manufacturers may choose to stop the production of certain medical devices. Furthermore, if certain medical devices do not get their certificates on time these products may become temporarily unavailable. Ask your suppliers to inform you in good time about the availability of the devices you need.”
About The Author
Leonard (Leo) Eisner is the principal medical device product safety and regulatory consultant at Eisner Safety Consultants. Known as the “IEC 60601 Guy,” he speaks around the world as an international expert on electrical safety and IEC 60601-1. Leo is involved in many IEC 60601 standards committees: IEC TC 62 / SC 62A: MT28 (Electrical Hazards), MT 30 (Overheating, fire protection and additional hazards), Ad Hoc Group 62368 (Sub-group of MT28 on feasibility of incorporating IEC 62368-1 into 60601-1), and WG14 (In charge of IEC 60601-1 Interpretations) that are working on the draft IEC 60601-1, 3rd edition + Amendment 2 project; is the US co-chair of IEC SC62D (majority of IEC 60601-2-xx and IEC/ISO 80601-2-xx standards); convener of IEC SC62D JWG9 (IEC/ISO 80601-2-58); member of IEC TC 62 / SC 62A , JWG6 (home care applications), JWG8 (EMS environment), and on the US TAG for TC62, SC62A, SC62D, AAMI Home Care & EMS environments, AAMI Electrical Safety (ES – develops ANSI/AAMI ES 60601-1) and AAMI Hospital Bed Working Group (EV/WG 05). He is a registered professional engineer in safety and has over 30 years’ experience in product safety. Leo manages the LinkedIn discussion group IEC 60601 Series – Medical Electrical Equipment. He can be reached at Leo@EisnerSafety.com.