
Closing The Loop On Risk Management With ISO 14971:2019
By Edwin Bills, Consultant
This is the second in a series of articles on the changes in the medical device risk management standard ISO 14971 that were released in December of 2019 and supported by guidance in the ISO TR 24971:2020 technical report or guidance on the medical device risk management standard. The standard and the technical report were reorganized and clarifications of the concepts of medical device risk management were included in the two documents. Few changes in the process resulted, but this article covers the major change in the process: production and post-production information covered in Clause 10 of the standard. Part 1 of this series covered the risk acceptability criteria and policy. The next and final article in this series will cover the topic of benefit-risk evaluation.
were released in December of 2019 and supported by guidance in the ISO TR 24971:2020 technical report or guidance on the medical device risk management standard. The standard and the technical report were reorganized and clarifications of the concepts of medical device risk management were included in the two documents. Few changes in the process resulted, but this article covers the major change in the process: production and post-production information covered in Clause 10 of the standard. Part 1 of this series covered the risk acceptability criteria and policy. The next and final article in this series will cover the topic of benefit-risk evaluation.
How Did We Get Here?
Risk management for medical devices developed over time, beginning with a European standard, EN 1441, on risk analysis, released in 1994. That led to an international risk analysis standard, ISO 14971-1, which was released in 1998. The developers of the international standard realized that there was more to the story than simply risk analysis, and immediately embarked on the development of a standard to manage the entire risk process, which became ISO 14971, Application of Risk Management to Medical Devices, first released in 2000. That standard defined what continues to be the management of the entire lifecycle of risk management for medical devices, including in vitro medical devices. One of the considerations discussed in development of the standard was the continuation of the process of risk management after the development of the medical device, especially after product release.
In its last clause, the 2000 edition of ISO 14971 covered closing the loop of the lifecycle with a section on post-production feedback of information. The clause was very short and included the responsibility of the manufacturer of the device to cover a post-production information evaluation step:
The information shall be evaluated for possible relevance to safety, especially the following:
a) if previously unrecognized hazards are present;
b) if the estimated risk(s) arising from a hazard is no longer acceptable;
c) if the original assessment is otherwise invalidated.
The next edition of the standard (2007) recognized that the process seemed to move from design and development to post-production, thus skipping the production phase, so the title was expanded on the last clause to “production and post-production” to indicate that there was information in the production phase that should be reviewed, similar to the post-production information. Users of the standard did not feel there was sufficient information to perform the requested activity. Reviews of standards are conducted every five years by ISO requirements; one of the comments by national committees during the vote on the 2000 edition’s final draft was to provide more guidance on how to implement this requirement. At that point, no further changes could be made due to ISO rules, so these comments were filed for the next edition.
In the vote to approve the final draft of the 2007 edition of ISO 14971, users again submitted comments asking for more information on the production and post-production information clause, among other subjects. Because of these comments and others, a project was begun to create a technical report to address the comments received in the voting process through a technical report instead of waiting for the next update to the standard. When the technical report on medical device risk management, ISO TR 24971:2013, was released, it included a section (three pages of guidance and a one-page flow chart) on production and post-production information among the areas addressed.
However, the technical report was not well promoted and very few people even recognized it was available. This problem probably occurred due to the development of the technical report after the standard, with no linkage between the documents. Following the vote to update the standard and the technical report in 2016, a team was assigned to specifically address these concerns. The team was to develop more information for the production and post-production phases of the device lifecycle, to address the regulatory postmarket surveillance requirements, and to align with ISO 13485:2016 requirements. As a result, the 2019 edition of ISO 14971 provides notes referring to the technical report in the standard, thus linking the documents.
ISO 14971:2019 Revision
Hopefully with the release of the 2019 edition of the standard, with 1.5 pages of requirements for this clause, and the 4.5 pages of referenced guidance in the ISO TR 24971:2020 technical report, the industry is more aware of the expanded requirements of the standard. It is important to understand that ISO 14971:2019 is identical to EN ISO 14971:2019 and that all the below discussion applies to compliance activities with either version of the standard.
Another reason to revise this clause was the expanded regulatory requirements of postmarket surveillance in the EU and the U.S. FDA. Postmarket surveillance is now the topic of an ISO technical report, ISO TR 20416:2020, which is referenced in ISO TR 24971. The technical report 20416 was created to bridge the ISO 13485:2016 requirements and the ISO 14971:2019 requirements and show how they addressed the regulatory requirements of postmarket surveillance that appeared in the MDR and FDA regulations and guidance. These regulatory requirements had led to the revision of ISO 13485 in its 2016 edition as well. Both of the standards, ISO 14971 and ISO 13485, have relied on the GHTF/SG3/N18:2010 Guidance on corrective action and preventive action and related QMS processes to provide direction in their revisions. More extensive information than in either of the standards is contained in the GHTF document, and it can be referenced for that additional information when developing or revising the risk management system and the quality management system. (The risk management system is covered in Clause 4 of ISO 14971:2019, and the concept was developed to meet the regulatory requirements in the new Medical Device Regulations [MDR]/In Vitro Diagnostic Regulation [IVDR] in Europe.)
In the 2019 edition, the flowchart that has been part of the standard since the beginning has been slightly revised, but not in the closing of the loop. The standard has always required that the information reviewed in the post-production phase be fed back to the beginning of the process for review and update of risk management process activities as required (See Figure 1 below).
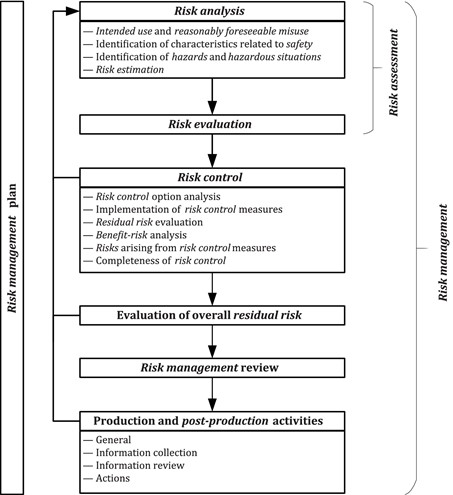
ISO 14971:2019 Figure 1: A schematic representation of the risk management process
The 2019 edition of 14971 now requires, in line with the regulatory requirements, an active process of:
- Information collection
- Information review
- Action (as required from the review).
The manufacturer can no longer sit by the phone and wait for complaints to come in.
ISO 14971:2019’s clause 10.1 states, “The manufacturer shall establish, document and maintain a system to actively collect and review information relevant to the medical device in the production and post-production phases” (emphasis mine). Notice the word “actively” here; the manufacturer must go seek information about the product and its risk(s), any new hazard(s), and any changes in “state of the art” rather than waiting around for complaints to come in. Other information sources should be reviewed in an active and ongoing process. The purpose is to assure that the risk management file contains accurate estimates of risk on the product and that the product risk be in line with the “state of the art” and the benefit for that particular device. Overall residual risk evaluation now includes benefit evaluation in all cases, not just when overall residual risk is considered unacceptable. ISO TR 24971:2020 provides 4.5 pages of guidance on the production and post-production phase of the device lifecycle, including a number of examples on developing a compliant process for this requirement of the standard.
Information Collection
ISO TR 24971:2020’s Table 7 contains examples of the information that manufacturers might collect from data sources, and it also contains examples of data elements within the data sources identified in ISO TR 24971. The table was developed from GHTF/SG3/N18:2010 Guidance on corrective action and preventive action and related QMS processes. Table 7, along with the Annex B Table in the GHTF CAPA guidance SG3/N18, will be very useful to you as manufacturer in developing information collection processes for ideas on what things to review in collecting information.
Information Review
It is interesting to note that in the 2019 edition of the standard, the aforementioned ISO 14971:2000 questions continue to drive the process with only slight revision:
The manufacturer shall review the information collected for possible relevance to safety, especially whether:
— previously unrecognised hazards or hazardous situations are present;
— an estimated risk arising from a hazardous situation is no longer acceptable;
— the overall residual risk is no longer acceptable in relation to the benefits of the intended use; or
— the generally acknowledged state of the art has changed.
(Emphasis mine.)
Note the incorporation of “benefit” in the analysis of overall residual risk. Overall residual risk is measured against benefit in all cases, and it is reflected in the post-production phase of the product lifecycle as well.
Action
The manufacturer has two requirements to meet in the action section:
- You must determine if the residual risk of the device is acceptable. Subsequently, you must determine if actions need to be taken to move the risk to acceptability both for the device already on the market and the devices currently in your control.
- You must ensure the risk management process itself is adequate. This activity relates to management review in ISO 13485 5.6, and the requirement in ISO 14971 4.2 for management to review the suitability of the risk management process.
Conclusion
Based on the extensive changes in Clause 10 in the standard, all manufacturers of medical devices and in vitro medical devices will need extensive review and possible changes to their production and post-production activities section of their quality systems in order to meet the new requirements of the risk management standard. While some may have started changes to their quality systems based on ISO 13485:2016 and the requirements there within Clause 8, “Measurement, analysis, and improvement,” those changes will probably be insufficient to meet the requirements of the new ISO 14971:2019. ISO TR 20416 illustrates the relationship between postmarket surveillance, quality management systems, and risk management systems.
You will need to meet the regulatory requirements in the new EU MDR and IVDR such as postmarket surveillance and postmarket clinical follow-up. The FDA has also developed a number of guidances on benefit-risk including the FDA guidance on postmarket actions, “Factors to Consider Regarding Benefit-Risk in Medical Device Product Availability, Compliance, and Enforcement Decisions,” issued at the end of 2016, containing useful considerations in developing both risk and benefit of a device, at least from FDA’s point of view. The ISO committee that created the revised ISO 14971:2019 tried to take these regulatory requirements into account in developing the revised standard, so implementation of the standard go a long way to meeting the regulatory requirements.
![Edwin Bills]() About The Author
About The Author
 About The Author
About The AuthorEdwin L. Bills has been a member of ISO TC 210 JWG1 for more than 20 years. This is the ISO group responsible for medical device risk management and the creation and maintenance of ISO 14971:2019, the risk management standard for medical devices, and ISO TR 24971:2020, the accompanying risk management guidance.