
FDA's Accreditation Scheme For Conformity Assessment (ASCA) Pilot Program Explained
By Mark Durivage, Quality Systems Compliance LLC
The U.S. Food and Drug Administration (FDA) Center for Devices and Radiological Health (CDRH) and Center for Biologics Evaluation and Research (CBER) recently posted for public comment The Accreditation Scheme for Conformity Assessment (ASCA) Pilot Program Draft Guidance for Industry, Accreditation Bodies, Testing Laboratories, and Food and Drug Administration Staff.
Biologics Evaluation and Research (CBER) recently posted for public comment The Accreditation Scheme for Conformity Assessment (ASCA) Pilot Program Draft Guidance for Industry, Accreditation Bodies, Testing Laboratories, and Food and Drug Administration Staff.
The Accreditation Scheme for Conformity Assessment (ASCA) Pilot Program Draft Guidance for Industry, Accreditation Bodies, Testing Laboratories, and Food and Drug Administration Staff, issued Sept. 23, 2019 seeks to use the FDA’s scientific resources more effectively and efficiently to protect and promote public health, as well as further encourage international harmonization of medical device regulations.
ASCA Pilot’s conformity assessment scheme will accredit testing laboratories that utilize FDA recognized accreditation bodies. Testing laboratories will be accredited using program specifications associated with each eligible standard and the requirements of ISO/IEC 17025:2017: General requirements for the competence of testing and calibration laboratories by recognized accreditation bodies.
The general ASCA Pilot conformity assessment scheme is as follows:
- Accreditation bodies may apply to participate in the ASCA Pilot
- FDA recognizes qualified for ASCA Pilot participation
- Testing laboratories receive accreditation from recognized accreditation bodies
- Testing laboratories may apply to participate in the ASCA Pilot
- FDA recognizes qualified testing laboratories for ASCA Pilot participation and grants ASCA Accreditation
- Device manufacturers may select ASCA-accredited testing laboratories for testing
- Device manufacturers may use summary test reports in premarket submissions from ASCA-accredited testing laboratories
FDA intends to periodically audit accreditation bodies and testing laboratories to ensure that they are adequately fulfilling program expectations.
ASCA Program Goals
The ASCA Pilot is intended to support FDA’s public health mission by providing increased confidence in testing results from ASCA-accredited testing laboratories, as well as potentially decreasing the burden of individual premarket submissions when manufacturers rely on testing completed by ASCA-accredited testing laboratories. The ASCA program goals are to:
- Enhance confidence in medical device testing
- Promote consistency and predictability in the premarket review process
- Encourage effective use of FDA resources
- Enhance regulatory efficiency
- Support international harmonization
ASCA Program Specifications
The ASCA Program has additional requirements beyond those required by ISO/IEC 17025. These additional requirements were developed with input from stakeholders at the public workshop titled “Accreditation Scheme for Conformity Assessment of Medical Devices to Food and Drug Administration-Recognized Standards.”
The ASCA ISO/IEC 17025 Sections with additional Program Specifications for the Biological Evaluation of Medical Devices are indicated in Table 1. For specific requirements, refer to Appendix A: ASCA Program Specifications for the Biological Evaluation of Medical Devices.
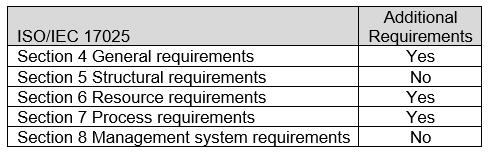
Table 1
The test methods included in the ASCA Pilot for Biological Evaluation of Medical Devices include:
- ASTM F756: Standard Practice for Assessment of Hemolytic Properties of Materials
- ISO 10993-4: Biological evaluation of medical devices – Part 4: Selection of tests for interactions with blood
- ISO 10993-5: Biological evaluation of medical devices – Part 5: Tests for in vitro interactions with blood cytotoxicity
- ISO 10993-10: Biological evaluation of medical devices – Part 10: Tests for irritation and skin sensitization
- ISO 10993-11: Biological evaluation of medical devices – Part 11: Tests for systemic toxicity
- USP <151>: Pyrogen Test
- ISO 10993-12: Biological evaluation of medical devices – Part 12: Sample preparation and reference materials
The ASCA ISO/IEC 17025 Sections with additional Program Specifications for the Basic Safety and Essential Performance of Medical Electrical Equipment, Medical Electrical Systems, and Laboratory Equipment are indicated in Table 2. For specific requirements, refer to Appendix B: ASCA Program Specifications for Basic Safety and Essential Performance of Medical Electrical Equipment, Medical Electrical Systems, and Laboratory Equipment.
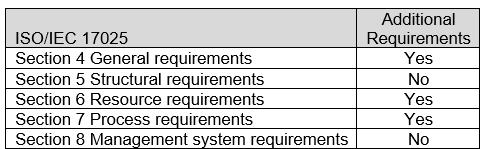
Table 2
The test methods included in the ASCA Pilot for Basic Safety and Essential Performance of medical devices and laboratory equipment include:
- ANSI/AAMI ES60601-1: Medical electrical equipment – Part 1: General requirements for basic safety and essential performance (along with the FDA-recognized collateral and particular standards in the 60601/80601 family)
- IEC 61010-1: Safety requirements for electrical equipment for measurement, control, and laboratory use – Part 1: General requirements (along with the FDA-recognized particular standards in the 61010 family)
Roles and Responsibilities
Accreditation bodies participating in the ASCA Pilot will accredit testing laboratories using the specifications of ISO 17025, plus ASCA program specifications, including the Biological Evaluation of Medical Devices and Basic Safety and Essential Performance of Medical Electrical Equipment, Medical Electrical Systems, and Laboratory Equipment as appropriate. Testing laboratories participating in the ASCA Pilot will perform testing in accordance with these same specifications.
Accreditation bodies, meanwhile, will need to modify their existing assessment tools to ensure compliance with the additional ASCA program specifications, including the Biological Evaluation of Medical Devices and Basic Safety and Essential Performance of Medical Electrical Equipment, Medical Electrical Systems, and Laboratory Equipment, as appropriate. Similarly, testing laboratories will need to perform a gap assessment to ensure compliance with the additional ASCA program specifications.
Device manufacturers may voluntarily choose to use a testing laboratory participating in the ASCA Pilot to conduct testing to be included in premarket submissions to FDA. The device manufacturer is responsible for the inclusion of appropriate device testing information in its premarket submission, as well as ensuring standards are selected and used appropriately, and that the declaration(s) of conformity provided in a premarket submission is consistent with Appropriate Use of Voluntary Consensus Standards in Premarket Submissions for Medical Devices.
FDA staff are responsible for ensuring consistent implementation of the processes and policies, as well as providing any training to ASCA Pilot participants necessary to maintain FDA’s confidence in the testing submitted by ASCA-accredited testing laboratories. Further, the FDA must ensure that accreditation bodies participating in the ASCA Pilot consistently meet its specified criteria for continued participation in the program.
FDA staff will conduct reviews of premarket submissions in accordance with existing statutes, regulations, and guidance. The FDA staff managing the ASCA Pilot are separate and independent from the FDA staff conducting premarket reviews.
Conclusion
Recent changes in organization, structure, and philosophy at the FDA are a positive sign for the medical device industry. This proposed new program is designed to make the regulatory submission process more efficient for the FDA and manufacturers.
I highly recommend that testing laboratories seeking to participate in the ASCA Program Pilot perform a comprehensive gap assessment to the additional requirements of ISO/IEC 17025 provided in Appendixes A and B of The Accreditation Scheme for Conformity Assessment (ASCA) Pilot Program Draft Guidance for Industry, Accreditation Bodies, Testing Laboratories, and Food and Drug Administration Staff prior to seeking accreditation from a recognized accreditation body.
The FDA requests that all comments on the draft guidance and program itself be submitted here no later than Dec. 23, 2019.
About the Author
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is Managing Principal Consultant at Quality Systems Compliance LLC, an ASQ Fellow and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including; CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is Managing Principal Consultant at Quality Systems Compliance LLC, an ASQ Fellow and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including; CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.