
How Do I Need To Update My QMS For FDA QMSR Amendment Compliance?
By Mark Durivage, Quality Systems Compliance LLC
 Now that the FDA has officially announced its intention to harmonize and modernize 21 CFR Part 820 Quality Management System Regulation (QMSR) for medical devices, the question is: What do I need to do to my quality management system (QMS)? If your QMS is based on the requirements of ISO 13485:2016, I suggest downloading 21 CFR Part 820 QMSR and analyzing where your current QMS may need updates to ensure compliance. I would recommend purchasing a copy of ISO 13485:2016 Medical devices - Quality management systems - Requirements for regulatory purposes and ISO 13485:2016 - Medical Devices - A Practical Guide, reading them, and performing a comprehensive gap analysis (assuming your current QMS was based on the requirements of 21 CFR Part 820).
Now that the FDA has officially announced its intention to harmonize and modernize 21 CFR Part 820 Quality Management System Regulation (QMSR) for medical devices, the question is: What do I need to do to my quality management system (QMS)? If your QMS is based on the requirements of ISO 13485:2016, I suggest downloading 21 CFR Part 820 QMSR and analyzing where your current QMS may need updates to ensure compliance. I would recommend purchasing a copy of ISO 13485:2016 Medical devices - Quality management systems - Requirements for regulatory purposes and ISO 13485:2016 - Medical Devices - A Practical Guide, reading them, and performing a comprehensive gap analysis (assuming your current QMS was based on the requirements of 21 CFR Part 820).
ISO 13485:2016 contains eight major clauses, including:
- Scope
- Normative References
- Terms and Definitions
- Quality Management System
- Management Responsibility
- Resource Management
- Product Realization
- Measurement, Analysis, and Improvement
Each clause contains the requirements as well as sub-clauses, which support the main clause by providing the details of the standard’s requirements for a QMS. For the purposes of this article, 21 CFR Part 820 will be referred to as the regulation and ISO 13485:2016 will be referred to as the standard. I will highlight the differences in each section of the standard and the procedures that will have to be modified to be compliant with the updated regulation, as well as the additional requirements necessary for the standard to be compliant with the regulation.
1. Scope
The standard applies to organizations that provide services including design and development, production, storage and distribution, installation, or servicing of a medical device and suppliers providing product, including QMS-related services. The regulation applies to finished devices and contract sterilization, installation, relabeling, remanufacturing, repacking, or specification development, as well as initial distributors of foreign entities that perform these functions. Organizations that manufacture parts or components of finished medical devices are not required to adopt the regulation but are encouraged to do so.
The FDA will still have the ability to grant exemptions or variances; however, that will not relieve the organization of following the requirements of the standards for the purposes of certification.
The FDA will still maintain inspectional jurisdiction over organizations regardless of their standard certification status. Additionally, the FDA will not issue certification to the standard or regulation. Generally, if an organization is not subject to the requirements of 21 CFR Part 807 Establishment Registration and Device Listing for Manufacturers and Initial Importers of Devices, the probability of the FDA inspecting your facility is negligible regardless of ISO 13585 certification status.
Modifications to the scope of the requirements include clarifications that conflicting regulations that are more specific take precedence only to the extent of the conflict.
The changes made here should not have any impact on the QMS documentation and are provided for clarification.
2. Normative References
ISO 9000:2015 Quality management systems - Fundamentals and vocabulary, the standard, and the regulation should be listed in Section 2, Normative References of the Quality Manual to aid auditors and inspectors during audits and official regulatory inspections.
3. Terms and Definitions
The terms and definitions found in ISO 9000:2015 Quality management systems - Fundamentals and vocabulary, the standard, and the regulation will apply. However, the definitions in the regulation will take precedence. These documents should be listed in Section 3, Terms and Definitions of the Quality Manual, along with any company specific terms, to aid auditors and inspectors during audits and official regulatory inspections.
4. Quality Management System
The failure to comply with any applicable requirement in the regulation renders a device adulterated under section 501(h) of the Federal Food, Drug, and Cosmetic Act. Such a device, as well as any person responsible for the failure to comply, is subject to regulatory action. This is in addition to any action that may by taken by the ISO certifying body.
The standard requires the organization to document a Quality Manual, which was not previously required by the regulation. The Quality Manual should describe at a high level how your organization complies with the standard, the regulation, and other applicable regulatory requirements. The Quality Manual should not be a regurgitation of the standard and the regulation. Any exclusions and non-applicabilites should be documented, with appropriate justification provided. A best practice is to include a compliance matrix with maps showing how and where your QMS addresses each requirement of the standard and the regulation to aid auditors and inspectors during audits and official regulatory inspections.
The regulation requires the manufacturer to document a QMS that complies with the requirements of the standard and the regulation and the following regulatory requirements as applicable:
- 21 CFR Part 803 Medical Device Reporting
- 21 CFR Part 806 Medical Devices; Reports of Corrections and Removals
- 21 CFR Part 821 Medical Device Tracking Requirements
- 21 CFR Part 830 Unique Device Identification
I suggest, as applicable, listing these additional regulatory requirements in Section 2, Normative References of the Quality Manual.
Your Control of Records standard operating procedure (SOP) must require the signature of everyone who approves or re-approves records (paper or electronic) and the date of the approval.
Your Control of Records SOP and/or External Inspections SOP should specify that records deemed confidential should be marked as such to aid the FDA in determining whether the information contained in the record may be disclosed to the public.
5. Management Responsibility
With the adoption of the standard, there will be a greater emphasis on identifying, analyzing, evaluating, controlling, and monitoring risk throughout the product life cycle to ensure that the devices are safe and effective. Management will be responsible for ensuring risk management and risk-based thinking are considered throughout the entire QMS, including planning, outsourcing, design and development, traceability, purchasing controls, acceptance activities, production and process controls, servicing, installation, analysis of data, and corrective and preventive actions (CAPA).
The regulation defines Top Management as those senior employees of a manufacturer who have the authority to establish or make changes to the manufacturer’s quality policy and quality management system. This position was previously referred to as management with executive responsibility. Update your Quality Manual and SOPs including Management Responsibility, Management Review, Reporting to Regulatory Authorities, etc., to reflect the use of the appropriate terminology, top management.
6. Resource Management
No specific differences exist between the requirements of the standard and the regulation, with the exception of the potential application of risk management and risk-based thinking.
7. Product Realization
Your Communications SOP should reference 21 CFR Part 806 Medical Devices; Reports of Corrections and Removals and how your organization will communicate with customers and the FDA in the event of a correction or removal as applicable.
Your Design and Development SOP will need to require the application of design controls to class II, class III, and certain class I devices. Class I devices are devices automated with computer software as well as the devices listed in the following table:
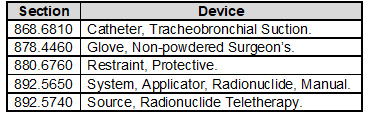
The regulation’s requirement for a design review requiring an individual(s) who does not have direct responsibility for the design stage being reviewed has not been transferred to the standard. However, you may want to consider adding this best practice to your Design and Development SOP.
Your Control of Production and Service Provision SOP will need to require the recording of the unique device identifier (UDI) for each medical device or batch of medical devices in the device history record (DHR).
You will need to update your Labeling and Packaging SOP to ensure labeling and packaging have been examined for accuracy prior to release or storage and, where applicable, to include the following:
- The correct UDI or universal product code (UPC), or any other device identification(s);
- Expiration date;
- Storage instructions;
- Handling instructions; and
- Any additional processing instructions.
The release of the labeling for use must be documented by obtaining the signature of everyone who approves the release (paper or electronic) and the date of the release.
Your Labeling and Packaging SOP must also ensure labeling and packaging operations have been established and maintained to prevent errors, including, but not limited to, inspection of the labeling and packaging immediately before use to assure that all devices have correct labeling and packaging, as specified in the medical device file. The results of labeling inspection must be documented by obtaining the signature of everyone who approves the release (paper or electronic) and the date of the release.
You will need to update your Servicing SOP to ensure the following information is recorded for servicing activities:
- The name of the device serviced;
- Any UDI or UPC, and any other device identification(s);
- The date of service;
- The individual(s) who serviced the device;
- The service performed; and
- Any test and inspection data.
Your Identification and Traceability SOP will need to document a system to assign a unique device identification to the medical device in accordance with the requirements of 21 CFR Part 830 Unique Device Identification.
You will also need to update your Identification and Traceability SOP. Traceability requirements from the standard for implantable medical devices will now additionally apply to devices that support or sustain life or for those for which the failure to perform, when properly used in accordance with instructions for use provided in the labeling, can be reasonably expected to result in a significant injury. Additionally, your Identification and Traceability SOP should reference 21 CFR Part 821 Medical Device Tracking Requirements and define the process for tracking such devices as applicable.
8. Measurement, Analysis, and Improvement
You will need to update your Complaint Handling SOP to ensure complaints that must be reported to the FDA as required by 21 CFR Part 803 Medical Device Reporting, complaints that a manufacturer determines must be investigated, and complaints that the manufacturer investigated regardless of those requirements capture, at a minimum, the following information:
- The name of the device;
- The date the complaint was received;
- Any UDI or UPC, and any other device identification(s);
- The name, address, and phone number of the complainant;
- The nature and details of the complaint;
- Any corrective action taken; and
- Any reply to the complainant.
Your Reporting to Regulatory Authorities SOP and/or Actions in Response to Nonconforming Product Detected After Delivery SOP should reference 21 CFR Part 806 Medical Devices; Reports of Corrections and Removals and how the organization will communicate with customer and the FDA in the event of a correction or removal as applicable.
Questions
If you have questions about the transition from 21 CFR Part 820 to ISO 13485:2016, please feel free to contact the author.
About the Author:
 Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.
Mark Allen Durivage has worked as a practitioner, educator, consultant, and author. He is managing principal consultant at Quality Systems Compliance LLC, an ASQ Fellow, and SRE Fellow. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. He holds several certifications including CRE, CQE, CQA, CSQP, CSSBB, RAC (Global), and CTBS. He has written several books available through ASQ Quality Press, published articles in Quality Progress, and is a frequent contributor to Life Science Connect. Durivage resides in Lambertville, Michigan. Please feel free to email him with any questions or comments.