
How To Build A Value-Added GMP Supplier Management Program
By Mark Durivage, Quality Systems Compliance LLC

Typically, supplier management programs use a supplier survey or an on-site audit to verify if a supplier is compliant with the requirements of a standard or regulation. However, for a supplier management program to provide added value to an organization, the supplier evaluation should look beyond the requirements of a standard or regulation. A value-added supplier management program should also consider items such as supplier capability, capacity, and contingency planning.

Requirements And Background
There are several International Organization for Standardization (ISO) standards, Food and Drug Administration (FDA) regulations, and national and international guidance documents that provide direction and lay out the framework for successfully implementing, maintaining, and sustaining an effective and robust quality management system, regardless of company type or size or the products and services it provides, requiring the use of risk-based thinking to manage suppliers. These include but are not limited to the following:
ISO 9001:2015 - Quality management systems — Requirements
8.4.1 The organization shall ensure that externally provided processes, products and services conform to requirements. The organization shall determine the controls to be applied to externally provided processes, products and services when:
a) products and services from external providers are intended for incorporation into the organization’s own products and services;
b) products and services are provided directly to the customer(s) by external providers on behalf of the organization;
c) a process, or part of a process, is provided by an external provider as a result of a decision by the organization.
The organization shall determine and apply criteria for the evaluation, selection, monitoring of performance, and re-evaluation of external providers, based on their ability to provide processes or products and services in accordance with requirements. The organization shall retain documented information of these activities and any necessary actions arising from the evaluations.
8.4.2 The organization shall ensure that externally provided processes, products, and services do not adversely affect the organization’s ability to consistently deliver conforming products and services to its customers. The organization shall take into consideration the potential impact of the externally provided processes, products, and services on the organization’s ability to consistently meet customer and applicable statutory and regulatory requirements
ISO 13485:2016 - Medical devices — Quality management systems — Requirements for regulatory purposes
7.4.1 Purchasing process requires the organization shall document procedures to ensure that purchased product conforms to specified purchasing information. The organization shall establish criteria for the evaluation and selection of suppliers. The criteria shall be based on the effect of the purchased product on the quality of the medical device and proportionate to the risk associated with the medical device.
21 CFR 820 - Quality System Regulation
820.50 Purchasing controls
Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements.
(a) Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants. Each manufacturer shall:
(1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality requirements. The evaluation shall be documented.
(2) Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results.
GHTF/SG3/N17:2008 - Quality Management System – Medical Devices – Guidance on the Control of Products and Services Obtained from Suppliers
3.1 Planning. In establishing the controls for product and services obtained from suppliers, it is expected that planning activities initiate the process. The output of this activity may be in the form of design and development plans, quality plans, purchasing plans, etc., as defined in the manufacturer’s QMS. The manufacturer should consider the objectives, risks, requirements, processes, and resources and demonstrate that effective controls are in place and regulatory obligations are met.
3.1.4 Identification of risk(s). As part of the planning activities, the manufacturer should identify the risks associated with the product or services to be obtained.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) - ICH Harmonised Tripartite Guideline Quality Risk Management Q9
II.5 Quality Risk Management as Part of Materials Management
Assessment and evaluation of suppliers and contract manufacturers
To provide a comprehensive evaluation of suppliers and contract manufacturers (e.g., auditing, supplier quality agreements).
Pharmaceutical Inspection Co-Operation Scheme (PIC/S) - Guide To Good Manufacturing Practice For Medicinal Products Part I
5.26. Starting materials should only be purchased from approved suppliers named in the relevant specification and, where possible, directly from the producer. It is recommended that the specifications established by the manufacturer for the starting materials be discussed with the suppliers. It is of benefit that all aspects of the production and control of the starting material in question, including handling, labeling, and packaging requirements, as well as complaints and rejection procedures are discussed with the manufacturer and the supplier.
Pharmaceutical Inspection Co-Operation Scheme (PIC/S) - Guide To Good Manufacturing Practice For Medicinal Products Part II
7.11 Manufacturers of intermediates and/or APIs should have a system for evaluating the suppliers of critical materials.
7.12 Materials should be purchased against an agreed specification, from a supplier or suppliers approved by the quality unit(s).
7.31 Supplier approval should include an evaluation that provides adequate evidence (e.g., past quality history) that the manufacturer can consistently provide material meeting specifications.
The above regulations, standards, and guidance documents refer to the requirements necessary to manage the supplier/purchasing controls function. However, to maintain an effective value-added supplier/purchasing controls function, one must look beyond the requirements of the regulations and standards.
Beyond The Regulations And Standards
Supplier management, including qualification, requalification, and performance monitoring, should also consider and assess items such as supplier capability, capacity, and contingency planning. The supplier management process should be thought of as a life cycle. Figure 1 provides an example of the phases in an effective supplier management program.

Figure 1: Example supplier management program
Qualification
The qualification of a supplier phase typically involves an assessment of a potential supplier’s quality management system (QMS) using a supplier survey, ISO certification review, or an on-site audit. During the qualification phase, many companies will rely on a third-party ISO certification. However, in my experience, this can be a very risky venture. Although ISO certification should indicate a supplier meets the requirement of a particular standard, there is much variation from registrar to registrar and from auditor to auditor. These inconsistencies should be minimized by auditing the supplier yourself to ensure the supplier meets your organization’s expectation for a given ISO standard (ISO 9001, ISO 13485, ISO 15378, ISO 17025, etc.). Relying on an ISO certificate may be acceptable for medium- or low-risk suppliers, but should never be acceptable for high-risk, critical, or key suppliers in the GMP-regulated industries.
When performing a supplier qualification audit for high-risk, critical, or key suppliers, the organization should focus on the items that are the most important for the products or services being procured. Figure 2 provides a high-level overview of the ISO 9001:2015 standard mapped to the plan-do-check-Act (PDCA) cycle.
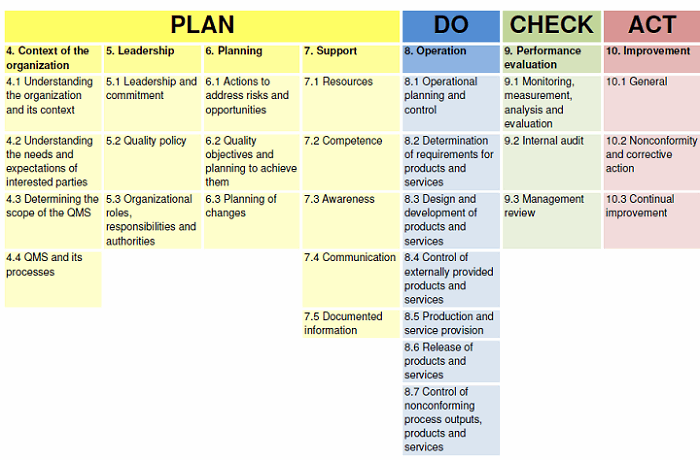
Figure 2: ISO 9001:2015 mapped to the PDCA cycle
For most supplier audits, I have prioritized my focus on the following activities of the PDCA cycle, in the following order:
1. Operation (Clause 8)
2. Support (Clause 7)3. Performance Evaluation (Clause 9)4. Improvement (Clause 10)
5. Planning (Clause 6)
6. Leadership (Clause 5)
7. Context of the Organization (Clause 4)
In a perfect world, the entire QMS should be evaluated during the audit. However, prioritizing the clauses is a practical reality. During a one-day audit, prioritizing as shown above will provide the most value using risk-based thinking. I have seen supplier audit reports where the auditor spent time worrying about whether the owner had the quality policy posted in their office, resulting in not having time to review the calibration program. The audit time would be better utilized assessing the supplier’s capability and capacity that directly impact the products and/or services being procured.
Supplier capability directly affects the goods and services being provided. The old adage "You can't make Swiss watch parts with blacksmith’s tools" highlights the importance of assessing a potential supplier’s ability to design, manufacture, inspect, and deliver quality products during an audit. While assessing the supplier’s capabilities, look for signs of investments in technology, including software, hardware, and equipment, as well as infrastructure and people. If investments have not been made, that is usually a sign of larger systemic issues that may affect your company’s needs.
Along with supplier capability, the supplier’s capacity should be assessed during an audit. From my supplier management and auditing experience, assessing supplier capacity is very difficult. Factors such as the supplier’s operating shifts, overtime policy, people (expertise), facility, availability of tooling, and availability of materials are generally good indicators of a supplier’s capacity. Figure 3 depicts a simple supplier gauge of capacity. Color-coding the factors using red, yellow, and green can help the auditor assess the supplier’s current and future capacity constraints.

Figure 3: Supplier capacity gauge
For example, if the supplier’s capacity is assessed as green, the supplier may be underutilized, possibly due to poor quality, or perhaps new capability has recently been introduced. If the supplier’s capacity is assessed as yellow, it may indicate there is good balance. From experience, I usually feel most comfortable selecting a supplier that is currently in the yellow zone. A supplier that falls in the red zone is a supplier to avoid. Although the red zone can be an indicator of superior quality, a supplier in the red zone may not have the ability to tackle a new project or grow with your organization’s needs. As you may have guessed, judging whether a supplier’s capability is red, yellow, or green is very subjective and requires a highly skilled auditor with expertise with the process and technologies under consideration.
Onboarding
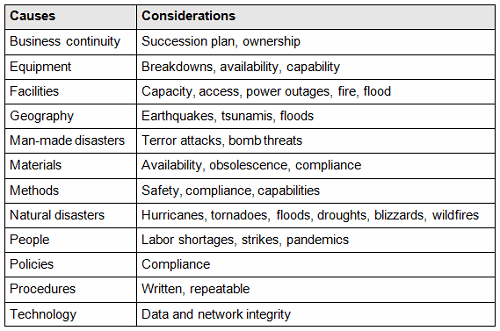
The supplier onboarding process, rather than the supplier qualification phase, is the time to articulate your requirements and expectations for supplier quality agreements, change notification requirements, capability expectations, capacity utilization, inspection expectations, validation requirements, and contingency planning above and beyond those in the applicable regulations and standards. Table 1 provides examples of items to consider including in a contingency plan.
Most companies will communicate and share the additional requirements and expectations through a supplier quality manual or other similar document. During the onboarding phase, supplier quality and purchasing will articulate any additional requirements and seek written acknowledgement, usually by signing a copy of the supplier quality manual. Sharing the supplier quality manual and obtaining official acknowledgement will help minimize future issues regarding requirements and expectations.
Supplier quality agreements and change notification requirements are generally negotiated, formalized, and executed during the onboarding process and are usually required before a purchase order is issued. There are many elements to consider during the generation of a supplier quality agreement, including but not limited to the following:
- Inspection requirements
- Process validation expectations
- Process monitoring requirements
- Agreement for auditing documents, records, and processes
- Agreement and deadlines for responding to quality issues
- Agreement to cooperate with investigations due to complaints, non-compliance reports (NCRs), and corrective and preventive actions (CAPAs)
Change notification requirements allow the organization to assess if a potential change will impact the goods or services being acquired. Change notification requirements generally include but are not limited to the following elements:
- Change in ISO scope
- ISO suspension
- Unfavorable FDA inspections
- Transfer to another registrar or agency in their registration, licensure, certification, or accreditation
- Change in ownership
- Change in the company name
- Composition any raw materials
- Change in the method of producing, processing, or testing
- Change in subcontractors for producing, processing, or testing
- Change of the manufacture site
Table 1: Example Contingency Plan Considerations
Development
The supplier development phase is probably the most time-consuming of the supplier management program. During this phase, design of experiments (DOEs), process optimization, and process validations may be necessary. There may be times, especially with smaller supplier sites (150 or fewer employees), when the supplier may not have the necessary technical expertise. In those cases, the organization may have to support technical development activities with quality, manufacturing, and processing engineering expertise or provide a consultant.
Monitoring
Supplier monitoring is an ongoing activity that usually includes metrics related to first pass yield (FPY), number of lots/parts received compared to the number of lots/parts rejected, and on-time delivery. Of course, there are many other metrics, but generally these are the easiest to access data to monitor. These metrics are usually summarized and reported to the supplier through a quarterly scorecard. The supplier scorecard can also be used to trigger on-site audits and other activities, including probationary status, restricting new orders, increased inspection requirements, and, in some cases, phasing out or desourcing the supplier.
Phase-out
The supplier management program generally ends when the parts or services are no longer required and when performance monitoring metrics have consistently yielded poor results. These are two discrete events. Phasing out a supplier for poor performance is not the same as desourcing a supplier when the parts or services provided are no longer required. The decisions made during this phase should be documented and include the rationale for the decision(s) made.
Conclusion
I cannot emphasize enough the importance of documenting the tools and methods used to implement a value-added supplier management program. The methods presented in this article can and should be utilized based upon industry practice, guidance documents, and regulatory requirements.
This article series has introduced other methods for integrating supplier management in the quality management system. The articles in the series include:
- Integrating Risk Management In The Quality Management System — A Primer
- An Introduction To qFMEA — A Tool For QMS Risk Management
- Using Risk-Based Thinking To Manage Suppliers
- Contingency Plans: An Essential Quality Management System Risk Tool
References:
- Durivage, M.A., 2017, The Certified Supplier Quality Professional (CSQP), Milwaukee, ASQ Quality Press
About The Author:
 Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, and connect with him on LinkedIn.
Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments, and connect with him on LinkedIn.