
MDSAP's Effect On The Internal Audit Process
By Mark Durivage, Quality Systems Compliance LLC
The Medical Device Single Audit Program (MDSAP) is recognized by officially approved Auditing Organizations (ISO  Registrars) to conduct a single regulatory audit, of a medical device manufacturer, that satisfies relevant requirements of the regulatory authorities participating in the program.
Registrars) to conduct a single regulatory audit, of a medical device manufacturer, that satisfies relevant requirements of the regulatory authorities participating in the program.
There currently are five country participants and two official observers involved with MDSAP, including:
- Australia’s Therapeutic Goods Administration (TGA)
- Brazil’s Agência Nacional de Vigilância Sanitária (ANVISA)
- Health Canada/Santé Canada
- Japan’s Ministry of Health, Labour and Welfare (MHLW) and Pharmaceuticals and Medical Devices Agency (PMDA)
- The United States of America’s Food and Drug Administration (FDA)
MDSAP also has two official observers; the World Health Organization (WHO) Prequalification of In Vitro Diagnostics (IVDs) Programme, and the European Union (EU).
A full MDSAP Audit comprises seven process chapters, including:
- Chapter 1 Process: Management
- Chapter 2 Process: Device Marketing Authorization and Facility Registration
- Chapter 3 Process: Measurement, Analysis and Improvement
- Chapter 4 Process: Medical Device Adverse Events and Advisory Notices Reporting
- Chapter 5 Process: Design and Development
- Chapter 6 Process: Production and Service Controls
- Chapter 7 Process: Purchasing
Each Chapter Process contains multiple audit tasks that are to be verified/confirmed during the audit. Each audit task references the applicable clause of ISO 13485:2016, in addition to specific regulatory requirements from the five participating countries. Throughout the audit, there is a focus on risk management and linkages between the processes.
Requirements And Background
Two applicable requirements in ISO 13485:2016 Medical devices - Quality management systems -Requirements for regulatory purposes can be significantly impacted by MDSAP:
0.1 General
Several jurisdictions have regulatory requirements for the application of quality management systems (QMS) by organizations with a variety of roles in the supply chain for medical devices. Consequently, this
International Standard expects that the organization:
— identifies its role(s) under applicable regulatory requirements
— identifies the regulatory requirements that apply to its activities under these roles
— incorporates these applicable regulatory requirements within its QMS
The definitions in applicable regulatory requirements differ from nation to nation and region to region. The organization needs to understand how the definitions in this International Standard will be interpreted in light of regulatory definitions in the jurisdictions in which the medical devices are made available.
8.2.4 Internal audit
The organization shall conduct internal audits at planned intervals to determine whether the quality management system:
a) conforms to planned and documented arrangements, requirements of this International Standard, quality management system requirements established by the organization, and applicable regulatory requirements;
5.6.2 Review input
l) applicable new or revised regulatory requirements
5.6.3 Review output
c) changes needed to respond to applicable new or revised regulatory requirements
The phrase ‘applicable regulatory requirements’ is mentioned five times in the requirements of these five clauses, indicating the importance of monitoring the regulatory landscape and auditing the QMS to ensure compliance with applicable regulatory requirements.
Internal Audit Program
When an organization commits to participate or is otherwise required, what happens to the internal audit program? The organization probably will develop an audit plan, checklist, and report based upon the seven MDSAP Chapters, along with the applicable country specific requirements. However, this plan could potentially cause several unforeseen issues. But why?
The answer is simple: MDSAP is currently recognized by the five countries listed above, but other country specific requirements probably will not be fully addressed using a MDSAP audit plan, checklist, and report. A good example of requirements that are not fully embedded and may be overlooked during an internal audit based upon MDSAP, are the Medical Devices Directive 93/42/EEC and In Vitro Diagnostic Device Regulation (EU) 2017/746.
The simple solution is to add the additional, non-participating country specific requirements to the audit plan, checklist, and report. However, a second option — to develop separate supplemental audit documents to address the additional non-participating country specific requirements— is, in my opinion, the best solution. This second method makes it easier for auditors to assess audit program compliance with non-participating country specific requirements.
A particularly valuable tool to aid in development of an internal audit plan, checklist, and report is a comprehensive compliance matrix. A compliance matrix is a method to ensure the requirements of a standard or regulation have been addressed, and to identify where they are addressed. I have seen compliance matrixes built where multiple standards have been ‘lumped’ together and mapped to the organization’s QMS.
However, best practice is to make a matrix for each standard or regulation to which the organization must comply. Additionally, a compliance matrix is a valuable tool to aid external auditors in the understanding of the QMS’ organization and structure.
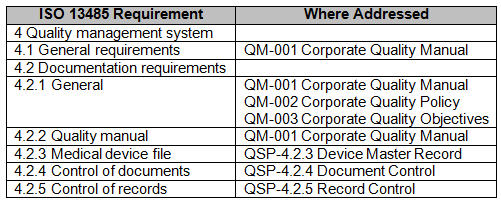
Fig.1 — Example partial Compliance Matrix for ISO 13485:2016
The management review process can also be affected by participating in MDSAP. Management review requires organizations to review applicable new or revised regulatory requirements. The management review process may be enhanced by listing and discussing the country specific regulations for locations where the organization designs, manufactures, labels, packages, distributes, installs, or services medical devices. A red-lined compliance matrix can be used to demonstrate the organizations’ review of applicable new or revised regulatory requirements.
Conclusion
Manufacturers will benefit by utilizing MDSAP, and there are no additional costs associated with participating in MDSAP. The manufacturer is able to choose any qualified Auditing Organization. Additionally, routine audits are scheduled and planned in cooperation with the auditing organization.
Further, there are no additional requirements (standards or regulations) with which manufacturers need to comply. In fact, MDSAP has mapped within the audit program:
- Medical device QMS requirements (ISO 13485:2016) and Quality System Regulation (21 CFR Part 820)
- QMS requirements of the Conformity Assessment Procedures of the Australian Therapeutic Goods (Medical Devices) Regulations (TG(MD)R Sch3)
- Brazilian Good Manufacturing Practices (RDC ANVISA 16/2013)
- Japan Ordinance on Standards for Manufacturing Control and Quality Control of Medical Devices and In Vitro Diagnostic Reagents (MHLW Ministerial Ordinance No. 169)
- Additional, specific requirements of medical device regulatory authorities participating in the MDSAP program within the audit program.
The goal is to provide management visibility to ensure QMS compliance with applicable new or revised regulatory requirements.
I cannot emphasize enough the importance of documenting (identifying and listing) country specific requirements. The approaches presented in this article can and should be utilized based upon industry practice, guidance documents, and regulatory requirements.
About the Author
 Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments.
Mark Allen Durivage is the managing principal consultant at Quality Systems Compliance LLC and an author of several quality-related books. He earned a BAS in computer aided machining from Siena Heights University and an MS in quality management from Eastern Michigan University. Durivage is an ASQ Fellow and holds several ASQ certifications, including CQM/OE, CRE, CQE, CQA, CHA, CBA, CPGP, CSQP, and CSSBB. He also is a Certified Tissue Bank Specialist (CTBS) and holds a Global Regulatory Affairs Certification (RAC). Durivage resides in Lambertville, Michigan. Please feel free to email him at mark.durivage@qscompliance.com with any questions or comments.
References:
- ISO 13485:2016 Medical devices—Quality management systems—Requirements for regulatory Purposes.
- Medical Device Single Audit Program (MDSAP) Companion Document 2017-01-06 MDSAP AU G0002.1.004