
Medical Device Postmarket Change Controls & FDA 510(k) Software Modification Guidance
By Yu Zhao, Rene Hardee, Randy Horton, Ashley Miller, Michael Iglesias, Roma Williams, and Jaden Maloney

The continued growth of Software as a Medical Device (SaMD) means that a large number of people are currently engaged in SaMD products. But there’s a smaller group of people who have successfully developed SaMD and gotten it to market.
What falls under Predetermined Change Control Plans (PCCPs), now that the 2023 Congressional Omnibus Bill calls for the FDA to implement them? We took a look at the Congressional language to evaluate software solutions beyond AI/machine learning (ML) that could benefit from a PCCP. In this three-part article series on postmarket change controls and implementing PCCPs for Class II SaMD products beyond AI/ML, we will discuss:
- Article 1 (this article): Medical Device Postmarket Change Controls & FDA 510(k) Software Modification Guidance
- Article 2: Deciphering New U.S. Laws Around Predetermined Change Control Plans
- Article 3: SaMD PCCP Implementation Beyond AI/ML: Considerations & Questions
Change Control Process In Six Steps For All Devices
One of the key defining characteristics of medical devices is that the device manufacturers will continue to make changes after the initial commercialization of the devices. Such postmarket changes can be planned or unplanned. They can be made for many reasons, including:
- to improve the device by adding new features or enhancing usability and
- to maintain or improve safety, effectiveness, quality, and/or reliability.
Examples of planned changes include, but are not limited to:
- expanding indications for use to include pediatric user populations,
- adding new product features,
- modifying the software user interface for better usability,
- adding a new supplier for a key component to improve supply chain management, and
- switching from manual to automatic production lines.
Examples of unplanned changes include, but are not limited to:
- switching to a new raw material supplier due to sudden bankruptcy of the existing supplier,
- implementing a security patch for a vulnerability newly identified by a third party,
- updating SaMD for a major operating system (OS) upgrade affecting Bluetooth communications, and
- starting identical production at a new manufacturing location after a strong earthquake has incapacitated the existing manufacturing facility.
The change control process is a key part of any medical device quality management system (QMS). To properly evaluate and implement any change made after the initial commercialization of the medical device, the change control process typically follows the six steps below.
- Understand the proposed change.
The device manufacturer needs to understand the scope and details of a proposed change and the reason for making such a change.
- Conduct an impact assessment on the change.
The device manufacturer needs to conduct a thorough impact assessment to understand the potential consequences of the proposed change on device safety, effectiveness, quality, and/or reliability, whether such change may potentially affect other hardware components or software modules in the same device or other connected external devices or systems, and any potential changes to the existing risk assessment.
Risk assessment of the change is one of the most critical parts of the impact assessment process. The manufacturer needs to review the existing risk management documents for the device and ask whether the proposed change could potentially affect the existing risk assessment. There are several ways that a proposed change could potentially affect the existing risk assessment, including:
- changing the severity and/or probability of occurrence of any existing harm, negatively or positively,
- introducing a new hazard or hazardous situation,
- making an existing risk control measure less effective, and
- requiring a new risk control measure.
To evaluate multiple changes (e.g., a component change with manufacturing process changes for component assembly), the impact assessment needs to be repeated for each proposed change and then for the cumulative effect of the changes combined.
- Perform appropriate verification and/or validation testing.
Based on the outcome of the impact assessment, the manufacturer needs to determine and perform an appropriate level of verification and/or validation testing, including bench testing, animal testing, usability testing, security testing, and human clinical study.
In addition to generating evidence for continued safety, effectiveness, device quality, and/or reliability, the verification and/or validation testing results may prompt the manufacturer to update the change impact assessment, including risk assessment, implement additional risk control measures, or revise regulatory assessment conclusion as described in Step 4 below.
- Conduct regulatory assessment and submit an FDA submission when appropriate.
For non-exempt Class II 510(k) devices and Class III PMA devices, the device manufacturer must determine whether a new FDA submission (including 510(k), or PMA including original PMA and various PMA supplements, excluding PMA Annual Report) is required to be submitted for the change based on the output of Step 2’s impact assessment and according to the relevant FDA regulations and guidance. In certain circumstances, the manufacturer could use the testing results from Step 3 to modify its FDA submission decision. In the case that a submission is required for the proposed change, the manufacturer will need to submit a new 510(k), PMA, or PMA supplement and wait for FDA clearance or approval prior to implementing the change.
If the regulatory assessment determines that a submission is not required, the manufacturer can proceed with change implementation after completion of change controls activities and documentation per its internal QMS requirements. Such regulatory assessment outcome is often referred to as document-to-file or letter-to-file.
For PMA devices, the manufacturer may determine that the potential impact of a change falls between a submission and a document-to-file, allowing the manufacturer to implement the change first, then notify the FDA by including such change in the next PMA Annual Report.
- Implement the change.
The manufacturer implements the proposed change according to its QMS requirements.
- Monitor the postmarket performance.
Following the implementation of the change, the manufacturer must continuously monitor real-world performance of the modified device and take appropriate action (such as failure mode analysis, corrective and preventive actions [CAPA], additional risk control, field safety notification, recall, etc.), according to its own QMS requirements.
Current FDA Guidance Regarding 510(k) Device Software Modifications
On Oct. 25, 2017, the FDA issued two updated guidance documents on 510(k) device modifications:
- Guidance for Industry and Food and Drug Administration Staff, Deciding When to Submit a 510(k) for a Change to an Existing Device1 (also known as “510(k) device modification guidance”)
- Guidance for Industry and Food and Drug Administration Staff, Deciding When to Submit a 510(k) for a Software Change to an Existing Device2 (also known as “510(k) software modification guidance”)
As our discussion focuses on Class II SaMD products, we provide a brief overview of the existing 510(k) software modification guidance, which was written before the introduction of the term “PCCP.” The guidance includes the following flowchart recommended for making regulatory assessment decisions on proposed software changes.
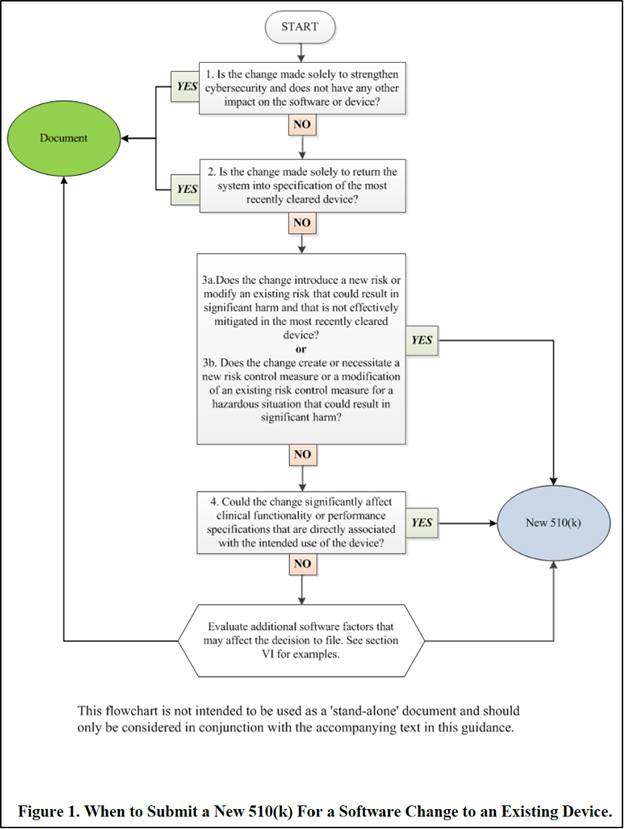
Figure courtesy of the FDA’s guidance document.
As illustrated in the flowchart and explained in detail in the accompanying text in the guidance, the questions in the flowchart are designed for the manufacturer to determine whether a 510(k) submission is required for the proposed software change. A 510(k) submission is required if one or both criteria is met:
- a major change or modification in the intended use of the device or
- a change or modification in the device that could significantly affect the safety or effectiveness of the device or result in a significant change or modification in design, material, chemical composition, energy source, or manufacturing process.
If, in addition to the proposed software change, there are other changes that may or may not be associated with the software change, or if the impact assessment concludes that the proposed software change may affect other aspects of the device (i.e., indications for use, labeling, or device performance, etc.), the 510(k) device modification guidance should also be used to determine whether a new 510(k) needs to be submitted to the FDA.
If the manufacturer determines that, based on the regulatory assessment, a new 510(k) is not necessary, then the manufacturer can write a document-to-file and implement the proposed change upon successful completion of the required verification and/or validation testing.
Stay tuned for the next articles in this series:
- Article 2: Deciphering New U.S. Laws Around Predetermined Change Control Plans
- Article 3: SaMD PCCP Implementation Beyond AI/ML: Considerations & Questions
References
About The Lead Author:
 Yu Zhao founded Bridging Consulting LLC, a consultancy dedicated to assisting AI startups and medical device companies in achieving innovation and regulatory compliance, in 2020. He has more than two decades of experience in the life sciences and technology space, and specializes in medical device regulatory, quality, and clinical affairs. His clients range from startups to large industry companies. During his tenure at Medtronic, he led regulatory affairs departments for multi-billion-dollar business units.
Yu Zhao founded Bridging Consulting LLC, a consultancy dedicated to assisting AI startups and medical device companies in achieving innovation and regulatory compliance, in 2020. He has more than two decades of experience in the life sciences and technology space, and specializes in medical device regulatory, quality, and clinical affairs. His clients range from startups to large industry companies. During his tenure at Medtronic, he led regulatory affairs departments for multi-billion-dollar business units.
Contributing authors are Rene Hardee, director of regulatory and program management at MedSec; Randy Horton, chief solutions officer at Orthogonal; Ashley Miller, director of regulatory affairs at Digital Diagnostics; Michael Iglesias, global quality advisor at Roche; Roma Williams, student at the University of Miami; and Jaden Maloney, student at Johns Hopkins University’s Whiting School of Engineering.